¶ 关于抗抑郁药
严重警告:抗抑郁药必须在专业医生指导下使用,禁止无处方擅自服药!
抗抑郁药并不是“安慰剂”或“提神剂”,而是一类作用于中枢神经系统、调节神经递质功能的处方精神药品。如果在没有医嘱的情况下擅自服用或更改剂量、随意停药,可能导致包括血清素综合征、激越发作、癫痫、致命性心律失常等严重后果,甚至危及生命。
与此同时,也请不要妖魔化这类药物。它们不是让人“上瘾”或“失控”的毒品,而是经由几十年全球科研人员不懈努力,逐步建立起的一整套有临床循证基础的抑郁治疗体系核心工具。如果你或你关心的人在服用这些药物,请尊重医生的判断,也尊重患者的选择。
¶ 人类最艰难的药学挑战之一
抗抑郁药的研发历史,几乎贯穿了整个现代神经科学的发展。自20世纪50年代偶然发现异丙肼(一种MAOI)具有改善情绪的作用以来,药理学家、精神病学家、生化研究员们持续在复杂的人脑神经递质网络中,寻找一把“重启心灵的钥匙”。
为了制造这种“可以改变情绪”的分子,人类耗费了数十年科研,投入了亿万资金。即使在今天,这些药物依然不是“精确制导”——但它们是我们在现阶段能给出最稳妥的选择。
同时,因为大部分抗抑郁药的时代久远,所以【历史背景】中的内容可能存在偏差,敬请谅解,Wiki主尽量为每一种药物发掘最原始的故事。
¶ 抗抑郁药的主流机制
大多数现代抗抑郁药,尤其是被最广泛使用的一类——选择性5-羟色胺再摄取抑制剂(SSRI),是通过让大脑中的血清素(5-HT)在神经之间停留更久,从而逐步缓解低落、焦虑、失眠、自责等症状。
这种作用不是立竿见影的。就像冬天修复结冰的道路一样,抗抑郁药需要几周时间让大脑慢慢“恢复通行”。但对许多人而言,它确实救了命。
¶ 抗抑郁药的迭代
| 代际 | 药物类型 | 特点 |
|---|---|---|
| 第一代:一线已淘汰 | MAOI(单胺氧化酶抑制剂) | 药效强,但食物药物禁忌多,副作用重,(一堆食物都不能吃,很容易一不小心吃死自己) |
| 第二代:完全淘汰 | TCA/TeCA(三环/四环类) | 药效稳定,但心脏毒性和抗胆碱副作用限制应用 |
| 第三代:临床一线 | SSRI(选择性5-HT再摄取抑制剂) | 安全性显著提升,成为临床一线 |
| 第三代延伸(“3.5代”):临床一线/二线 | SNRI(5-HT+NE再摄取抑制剂)、NDRI(NE+DA)、NaSSA 等 | 多机制复合型,适配不同病型的抑郁症 |
| 第四代:在研/有限使用 | NMDA受体拮抗剂、5-HT₁A/₂C调节剂、多通路新机制药物 | 代表药物如艾司氯胺酮(S-ketamine)、Auvelity,尚未普及 |
这些迭代背后,每一款药物都是无数科研工作者在动物实验、分子筛选、双盲临床试验中不断试错换来的。他们中许多人一生只为一款分子工作,也未必能看到它最终上市。
¶ 本篇的视角与目的:为MTF群体的抑郁治疗提供更匹配的参考
与其他常见的精神类Wiki不同,本篇在介绍各类抗抑郁药物时,将特别以跨性别女性(MTF)群体为核心视角,在每一款药物的解说中,加入对MTF人群可能产生的额外获益或潜在风险的分析。这种角度并不是为了强调差异化的特殊性,而是因为在现实精神科就诊体系中,MTF患者的生理结构、性激素状态、精神应激背景,与顺性别患者确实存在显著不同,而这恰恰常常被临床忽略。
当前中国精神卫生系统面临着极大的资源不平衡问题。一方面,全国每十万人仅有个位数合资格的精神科医生,真正具备抑郁、焦虑、双相等障碍精准识别与分型能力的临床人员更是凤毛麟角;另一方面,患者数量却在持续上升,尤其是创伤背景复杂、性别认同特殊、长期遭遇歧视的群体,如MTF,更易发展出慢性化、复合型精神障碍。
在如此资源匮乏的背景下,大量精神科医生不得不“快诊式”开药,使用有限的几种药物模板试图笼统覆盖不同病型的患者。这不仅因为医生难以抽出精力个别评估,更因为药房本身的供应系统受限,存在“某些药必须清库存”“医保优先使用品种”的现实操作问题。于是,大量患者——包括MTF人群——在完全不了解自身病理特征、药物机制、适配度的情况下,被动地接受一种“通用化”但并不真正适合自己的治疗方案,这些不是医生们的错,是社会复杂的现实。
更令人担忧的是,由于对精神疾病的污名化与医疗资源的信任危机,许多MTF患者选择沉默地承受不适、私自加减剂量,或干脆中断用药。这些行为都可能在不知不觉中加重病情,延长恢复周期,甚至诱发自杀风险。
因此,本篇的一个重要目的,是试图用通俗、系统、温和而不妥协的语言,帮助MTF群体理解:自己现在正在服用的药物是什么,它的原理是什么,它是否真的适合你,它是否可能正在延误你的康复,而你是否可以合理地、专业地去向医生申请换药。
精神类药物的调配,从来都不该是“试试看”,尤其是SSRI药物存在不同亚型靶点、以及有相当一部分占比的人对SSRI药物有受体不应答问题。(说人话就是这些人天生吃SSRI没用)它必须基于对自身状态的真实理解。而当医生的时间被压缩、药房的资源受限时,我们希望这篇文章能为你提供一个补足医生视野的信息起点。
我们深知,每一位MTF抑郁症患者都不是脆弱的,而是经历了超越常人数倍的情绪负荷与社会挑战之后,依旧选择活着的坚韧个体。你的情绪痛苦不该被忽略,你的药物不该被敷衍地对待。
如果你已经走到了服药这一步,那么请允许自己继续往前走一步——去理解它、质疑它、和它合作,而不是被动服从它。希望你能从本篇中获得真正属于你自己的选择权。
¶ 关于本篇的立场说明
本篇文章将对目前中国与国际主流使用的抗抑郁药物,逐一进行评述与归类。我们会尝试用浅显易懂的语言解释其机制、效果、副作用与适用人群,也会作出一定程度上的主观评价,以便读者快速理解药物之间的差异。
但请谨记:本篇所有评分和评论均基于现代药物体系中“横向对比”的视角,绝不意味着某款药物“无用”或“过时”。每一款药的存在,都是它在某个历史节点救过某些人。
抗抑郁药,是人类药学史上最难研发的品类之一,它不作用于肝脏或肾脏,而作用于意识、情绪与人格的化学基础。因此,它也应当被给予最谨慎的理解,和最温柔的尊重。致敬每一位让人类不再沉默于悲伤的科研人员。
¶ 名称须知:化合物名 ≠ 商品名
在阅读本篇Wiki或与他人交流抗抑郁药物时,请注意药品命名方式中可能存在的混淆。
每一种药物通常都会同时存在两种常见称呼:
化合物名称(Generic Name):这是该药物的有效化学成分名称,例如“氟西汀”、“艾司西酞普兰”、“舍曲林”;
商品名称(Brand Name):这是药厂在上市销售时所取的商品名,例如“百忧解(Prozac)”、“乐友(Lexapro)”、“左洛复(Zoloft)”等。
在本Wiki中,我们会统一使用“化合物名称”作为正文描述的主体,因为不同国家、不同地区、甚至不同厂家的商品名可能不同,而化合物名称更能指向药物的核心机制与特性。
同时,你也可能在药品说明书或包装盒上看到“盐酸舍曲林”、“草酸艾司西酞普兰”、“氢溴酸西酞普兰”这样的前缀名词。这些“草酸”、“盐酸”、“氢溴酸”等其实是药物制剂中的“盐形式”,它们不是药物本身的名字,而是化学稳定性与溶解度优化过程中的必要成分,属于“药物载体”或“辅料”范畴。
换句话说:
“草酸艾司西酞普兰”中的“艾司西酞普兰”才是关键;
“盐酸舍曲林”中的“舍曲林”才是发挥作用的有效成分。
因此,在向他人描述用药时,请尽量避免说“我在吃盐酸”“我吃的是草酸”这类表述,因为它们无法准确传达你所服用的药物种类,也可能导致误解。
¶ 盐形式
药品的有效成分大多数是有机碱(例如许多精神类药物都是胺类化合物,有“碱性”),而这些碱性化合物通常不容易稳定存在或溶解,因此在制药工业中会将它们与某种酸反应,形成盐。
这就类似于:
舍曲林(sertraline,本身是碱) + 盐酸(HCl) → 盐酸舍曲林(sertraline hydrochloride)
这样生成的“盐”形式稳定性更好,更容易溶于水,更适合做成片剂、胶囊、注射液等药物制剂。
所以你看到的“盐酸舍曲林”、“草酸艾司西酞普兰”、“氢溴酸西酞普兰”中的前缀(盐酸、草酸、氢溴酸)其实都是:
和药物反应生成“盐”的酸性配体;
它们只是药物的制剂形式,不是药物发挥作用的“本体”。
因此我们说:这些是“盐形式”,而不是“酸形式”。
为了避免混淆,本Wiki也会在每款药物条目中注明它的通用名、商品名以及常见盐形式,供查阅参考。
¶ 关于【原研】与仿制药以及价格的说明
在本Wiki中,药品的规格与厂家信息部分,部分药物前会标注【原研】字样,用以指代该药物由首个完成研发、上市并持有原始专利权的制药企业所生产的版本,俗称原研药。
原研药通常由大型跨国制药公司开发,研发周期长、投入资金巨大,价格也较为昂贵。而在其专利期届满后,其他厂家可以依法生产与其有效成分、剂量、给药方式一致的版本,这类药品被称为仿制药。
需要特别说明的是:
仿制药并不意味着质量差。经国家药监部门批准并通过一致性评价的仿制药,其在疗效与安全性上应当与原研药一致;
临床实际使用中,仿制药已被广泛替代原研药,尤其在医保范围内的药物分发体系中;
但也确实存在个别患者因辅料差异、代谢差异或心理预期等原因,认为不同厂家生产的版本“体感不同”,这类情况建议与医生讨论后再作更换。
因此,在本页面后续出现“【原研】”标记时,仅表示该药为最早上市版本,并不代表疗效更优,也不代表本Wiki对其推荐等级上的偏向。我们鼓励患者在了解药物来源的同时,理性看待原研与仿制的区别。
价格部分,Wiki主所在省份为云南,所以所有的药物价格均采用云南省药物采购挂网价格,不同省份间药物价格会有略微差距,但不会相差太多。
接下来的内容就是每一种抗抑郁药的详细数据了,可在右侧选择你想要跳到的段落进行查阅。
在写SSRI前,我很想先写一款药物:在第一代抗抑郁药前的“第零代”抗抑郁药,异丙烟肼。
¶ Generation 0:异丙烟肼(Iproniazid)
¶ 为什么称为“第0代”?
在本Wiki中,我们将异丙烟肼(Iproniazid)归类为“第0代抗抑郁药”,并非因为它是药理上最早的抗抑郁机制分子,也并非因为它是系统性设计用于治疗抑郁症的化合物,而是出于历史严谨性和源头追溯的考虑。
异丙烟肼的出现,是抗抑郁药物发展史上的一个偶然转折点。它最初被开发用于治疗肺结核,在1950年代的美国与欧洲临床中广泛使用。然而医生们惊讶地发现:接受异丙烟肼治疗的部分结核病人出现了情绪高涨、精力恢复、社交变得活跃的现象。起初这被当作“副作用”记录,但在精神科领域逐渐引发兴趣。
随后的研究发现,异丙烟肼具有抑制单胺氧化酶(MAO)的能力,从而提升大脑中血清素、去甲肾上腺素、多巴胺的浓度。这一发现不仅催生了后续MAOI类抗抑郁药的研发(如苯乙肼、司来吉兰等),更重要的是,它第一次将“神经递质”的生物化学调控与“情绪”之间建立了明确联系,由此催生出“单胺假说”——即抑郁症可能源于脑内某些神经递质水平的失衡。
因此,异丙烟肼虽然不是为抑郁症研发、且早已因肝毒性等副作用退出市场,但它毫无疑问是现代精神药物学的“第一个破局点”。我们将其独立列为“第0代抗抑郁药”,是对这个偶然发现给予足够的科学尊重,也作为通往后世数百种抗抑郁药的源头注脚。(可以理解为,异丙烟肼是一个偶然性发现的MAOI,而司来吉兰等药物是专门目的性开发的MAOI,因此异丙烟肼属于第0代抗抑郁药,或“第0号抗抑郁药”)
¶ 为什么要讲述异丙烟肼?
本Wiki不仅是抗抑郁药物的用药参考工具,更承担着追溯、记录、解释抗抑郁药发展历程的使命。而任何一个系统性的科学体系,都必须认真对待自己的起点。
异丙烟肼的故事代表着抗抑郁药物研究中最原始的逻辑转向——它不是对症设计的产物,而是被患者行为意外暴露出来的机制碎片。这个过程也提醒我们,科学的发展往往不是线性展开的,而是从误解、偶然、甚至副作用中逐渐拼凑出体系。药物科研,从本质上属于迭代性科技。
异丙烟肼以及MAOI大类已全部退出临床一线治疗方案,将异丙烟肼放入Wiki,不是重用该药物,更不是对其临床风险的忽略,而是为了让每一位读者明白:
在我们今天使用的SSRI、SNRI、NDRI、NMDA类抗抑郁药诞生之前,一切从一个治疗肺结核失败的副作用开始。我们要对所有的药物尤其是精神类药物抱有敬畏之心。
¶ SSRI:选择性血清素再摄取抑制剂
¶ 简介
选择性血清素再摄取抑制剂(SSRI) 是目前临床最常见的抗抑郁药类型之一,属于第三代抗抑郁药。它们通过调节大脑中的血清素(5-HT)水平来起效,被广泛应用于抑郁症、焦虑障碍、强迫症等疾病。
术语解释如下:
选择性:药物只作用于血清素系统,而不显著影响去甲肾上腺素、多巴胺等其他神经递质,从而副作用较少。
血清素:一种能影响情绪、睡眠、食欲等的神经递质,被认为与“快乐感”相关。
再摄取:神经元释放出的血清素会被“回收”进神经元,这是正常代谢过程。
抑制剂:即阻止这种“回收”的过程,让血清素在神经元之间停留更久,从而增强情绪调节。
把它们合到一起就是: 选择性 地只让 血清素 的 再摄取 (回收)环节给 抑制 掉的 药剂。
¶ 中国临床常用的一线 SSRIs
目前在中国最常用于治疗抑郁症的一线 SSRI 药物包括:
氟西汀(Fluoxetine)
帕罗西汀(Paroxetine)
舍曲林(Sertraline)
氟伏沙明(Fluvoxamine)
西酞普兰(Citalopram)
艾司西酞普兰(Escitalopram)
其中,舍曲林 和 艾司西酞普兰 是中国精神科临床最常开具的 SSRI,特别是在未成年患者中,舍曲林因其安全性更高而被优先使用。
以下排名为主观观点打分的,仅供参考。
| 排名 | 药物名称 | 常规群体评分 |
|---|---|---|
| 1 | 氟伏沙明 | 9.0 |
| 1 | 伏硫西汀 | 9.0 |
| 3 | 艾司西酞普兰 | 8.5 |
| 4 | 帕罗西汀 | 7.0 |
| 5 | 舍曲林 | 6.5 |
| 6 | 氟西汀 | 6.0 |
| 6 | 西酞普兰 | 6.0 |
| 排名 | 药物名称 | HRT群体评分 |
|---|---|---|
| 1 | 氟伏沙明 | 9.0 |
| 2 | 伏硫西汀 | 8.0 |
| 3 | 艾司西酞普兰 | 7.0 |
| 4 | 氟西汀 | 5.5 |
| 5 | 帕罗西汀 | 5.0 |
| 5 | 西酞普兰 | 5.0 |
| 7 | 舍曲林 | 4.5 |
¶ 氟西汀(Fluoxetine)LILLY-110140 【HRT群体不作为首要推荐用药,原因参考下方评价】
¶ 基本信息
药物国际通用名/商品名:氟西汀 / 百忧解(Prozac)
化学分子式 C17H18F3NO
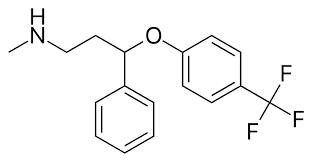
类别:选择性血清素再摄取抑制剂(SSRI)
NMPA核准适应症:重度抑郁症、强迫症、神经性暴食症、惊恐障碍
厂家/规格/售价:
常州四药制药有限公司 盐酸氟西汀片 10mg/14片 价格:20.46元
【原研】PATHEON FRANCE (礼来苏州制药有限公司) 盐酸氟西汀胶囊 20mg/28粒 价格:162.05元 (PATHEON FRANCE是LILLY的分包商,其中的药物本身生产来自LILLY原厂,广义上属于原研药)
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点受体作用机制 | 抑制血清素(5-HT)再摄取,增强突触间信号传递 |
| 亲和力/选择性 | 对血清素转运体有高度亲和力,其他递质作用极小 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 原药 4-6 天;活性代谢物去甲氟西汀 4-16 天 |
| 起效时间 | 1-2 周初效,4-6 周达到稳态 |
| 起效周期 | 需连续服药数周 |
| 首过效应/吸收 | 生物利用度高,代谢稳定 |
| CYP 代谢信息 | CYP2D6 主要参与,注意药物相互作用 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 缓解速度 | 起效相对缓慢,患者需有耐心 |
| 起效后表现 | 情绪稳定、兴趣改善、进食/睡眠恢复 |
| 精神症状适应性 | 对强迫症、暴食症疗效确切 |
¶ 副作用
中枢神经系统:失眠、头痛、激越、焦虑短期加重
性功能与体重:性欲减退、性功能障碍;部分患者体重变化明显
特殊副作用:罕见诱发躁狂发作、QT 延长、癫痫
注意:青少年使用氟西汀时可能有轻度自杀观念升高的风险,需密切监测情绪变化。
¶ 优劣情况
优势:半衰期长,停药撤反较轻,对暴食症、强迫症有额外适应性。
劣势:起效慢,性副作用突出,初期可能激越。
¶ 对于 MTF 群体的优劣情况
明显优势:部分患者报告焦虑控制能力提升,早期激动性减轻
明显劣势:性副作用可能加重本就受激素水平影响的性功能困扰,影响情感认同体验
¶ 用药注意
剂量:成人初始常用剂量为每日 20mg,可按需逐步调整,最大不超过 80mg。
联用情况:慎与苯二氮卓类合用,注意 QT 风险。
撤药建议:至少一个月的清除时间。
¶ 历史研发背景
氟西汀(Fluoxetine)是人类医学史上最早被合成、第二个上市的选择性血清素再摄取抑制剂(SSRI),它的出现标志着抗抑郁药物研究进入了全新的时代。作为SSRI家族的“始祖”之一,氟西汀不只是一个新药的名字,更是整个精神病学研究范式转换的标志性产物——它将抑郁症的治疗从“多胺综合提升”模式彻底引导至“血清素主导”路径,为后续几代抗抑郁药物奠定了理论基础。
20世纪50-60年代,抗抑郁药的研发主要聚焦在提升单胺类神经递质(如多巴胺、去甲肾上腺素、血清素)的整体浓度,主流药物包括三环类抗抑郁药(TCAs)和单胺氧化酶抑制剂(MAOIs)。这类药物机制粗放,影响多种神经递质,同时伴随显著的副作用。
然而,1966年前后,神经科学界开始尝试从中“找出罪魁祸首”:哪一种神经递质最关键?
研究者发现:无论是三环类还是MAOI类药物,它们无一例外都会影响血清素系统。
与此同时,动物实验中也观察到:当动物脑内的血清素浓度升高时,其攻击性显著下降——这种温和、镇静、非冲动性的行为改变,与抑郁症典型临床症状高度契合。
一个偶然的启发也来自嬉皮士文化中滥用的LSD(麦角酸二乙酰胺)。科学家发现,LSD作为一种强烈的致幻剂,能通过激活5-HT受体系统,引发精神状态剧烈变化,但它同样也使使用者出现情绪迟钝、进攻性削弱的现象。这进一步暗示了:血清素可能是调节情绪、攻击性、社会行为的关键介质。
最终,在1967年,有人首次提出:在抗抑郁研究中,血清素的角色比去甲肾上腺素(NE)更重要。到1969年,这一假说被具体化为“抑郁症源于大脑突触间隙中的血清素水平下降”的理论。尽管当时该理论并未获得广泛共识,却于同年1月18日,正式发表在《柳叶刀》杂志第132-136页上,引发全球药企和研究机构的高度关注,开始了对“血清素选择性靶点”的专项开发。
此时的礼来制药公司(Eli Lilly)在研究一种广泛使用的抗组胺药物 苯海拉明(Diphenhydramine),其结构属于单胺能再摄取阻滞剂,具有弱的血清素转运蛋白亲和力。科研人员发现,苯海拉明除了抗组胺特性外,还意外表现出对5-HT再摄取的轻微抑制作用。
这成了突破口。礼来的研发团队大胆推断:如果保留血清素转运选择性,同时完全去除抗组胺活性,是否能筛选出更专一、更安全的抗抑郁新药?
于是,他们将苯海拉明结构拆分为三段——左端、中心氧桥、右端,以氧为基点重新排列,删除H1受体活性片段,保留对5-HT转运蛋白的高选择性结构。进一步地,他们在苯环上引入了三氟甲基(-CF₃)基团以增强脂溶性和脑穿透力,同时优化药物的缓释能力和代谢稳定性。
(苯海拉明)
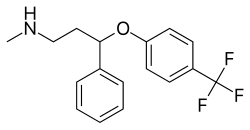
(氟西汀)
最终,1972年,氟西汀这一化合物被首次成功合成。
在长达十余年的临床验证与药理筛查后,氟西汀以“Prozac / 百忧解”之名于1987年正式上市。它不仅是第一款广泛流通、验证有效、安全性可控的SSRI,也真正让“选择性血清素再摄取抑制”从理论变为现实。
与此前“粗暴提神、副作用满溢”的老药不同,氟西汀以温和、渐进、较低副作用著称,改变了公众对“精神病药就是毒药”的刻板印象,并成为整个90年代抗抑郁治疗的代名词。此后,帕罗西汀、舍曲林、西酞普兰、艾司西酞普兰等相继面世,构成如今SSRI家族的完整谱系。
氟西汀的诞生并非偶然,它是精神病学从暴力控制走向神经递质精准调控的开端。正是这氟西汀,改变了整个精神科药物开发的方向,从“全谱激活”转为“单轴调控”,奠定了现代抗抑郁治疗的基本模型,也让“抑郁症是可治疗的神经递质失衡”逐步被大众接受。
如果要为“精神药物现代史”立一个开端碑,那一定是氟西汀。
中国批准时间:90年代中期引入国内市场
专利到期:2001年8月,随后出现大量仿制药
¶ 主观评分: 6/10(常规群体) 5.5/10(HRT群体)
¶ 主观评价:氟西汀作为最OG的SSRI起源药,它的功劳不可埋没,不过在2025年的抗抑郁药选择市场里,我主观给到的分数是6/5.5。首先药效方面氟西汀是中游偏上水平的,0.81nM中位数的SERT-Ki,对血清素转运体亲和力很强了,但相较于其他后来的SSRI,氟西汀存在几个问题:1.氟西汀的药物半衰期太太太长了,原型2-4天,活性代谢物能到7-15天甚至更久,硬说好处吧就是漏一两天不吃都没事,但是撤药和换药都太慢了,血药浓度下降速度慢到离谱,一般换药期间清除需要至少1个月才能确保安全,相比之下像是艾司西酞普兰的半衰期在24小时以内,差距巨大。 2.双相情感障碍群体不适用,氟西汀很容易转躁狂,因为情绪激活性太强了,有一点用力过猛内味儿,而且青少年使用氟西汀会增加自杀风险,我个人认为这是因为合成时引入三氟甲基导致的。 3.氟西汀的CYP2D6抑制能力太强了,很容易影响其他联用药物的代谢,血药浓度会出问题,HRT类药物的影响不太明确但是我觉得还是会有的,所以虽然CYP3A4方面氟西汀没什么问题,但仍然不是很建议MTF群体优先选择氟西汀。 综合来看,氟西汀现在比较适合的是无情绪、情绪低沉型的成年MDD(重度抑郁症)患者,而广泛性的抑郁、焦虑障碍等情况,不太适合选择氟西汀,如果你吃氟西汀感觉到莫名的狂躁、冲动、坐不住容易激的话…那就对咯,氟西汀不适合你,向医生申请花一个月时间断药,然后换其他药物吧。
¶ 舍曲林(Sertraline)CP-51974-1 【HRT群体不作为首要推荐用药,原因参考下方评价】
¶ 基本信息
药物国际通用名/商品名:舍曲林 / 商品名包括 Zoloft(原研)、乐友(国内)等
化学分子式C₁₇H₁₇Cl₂N
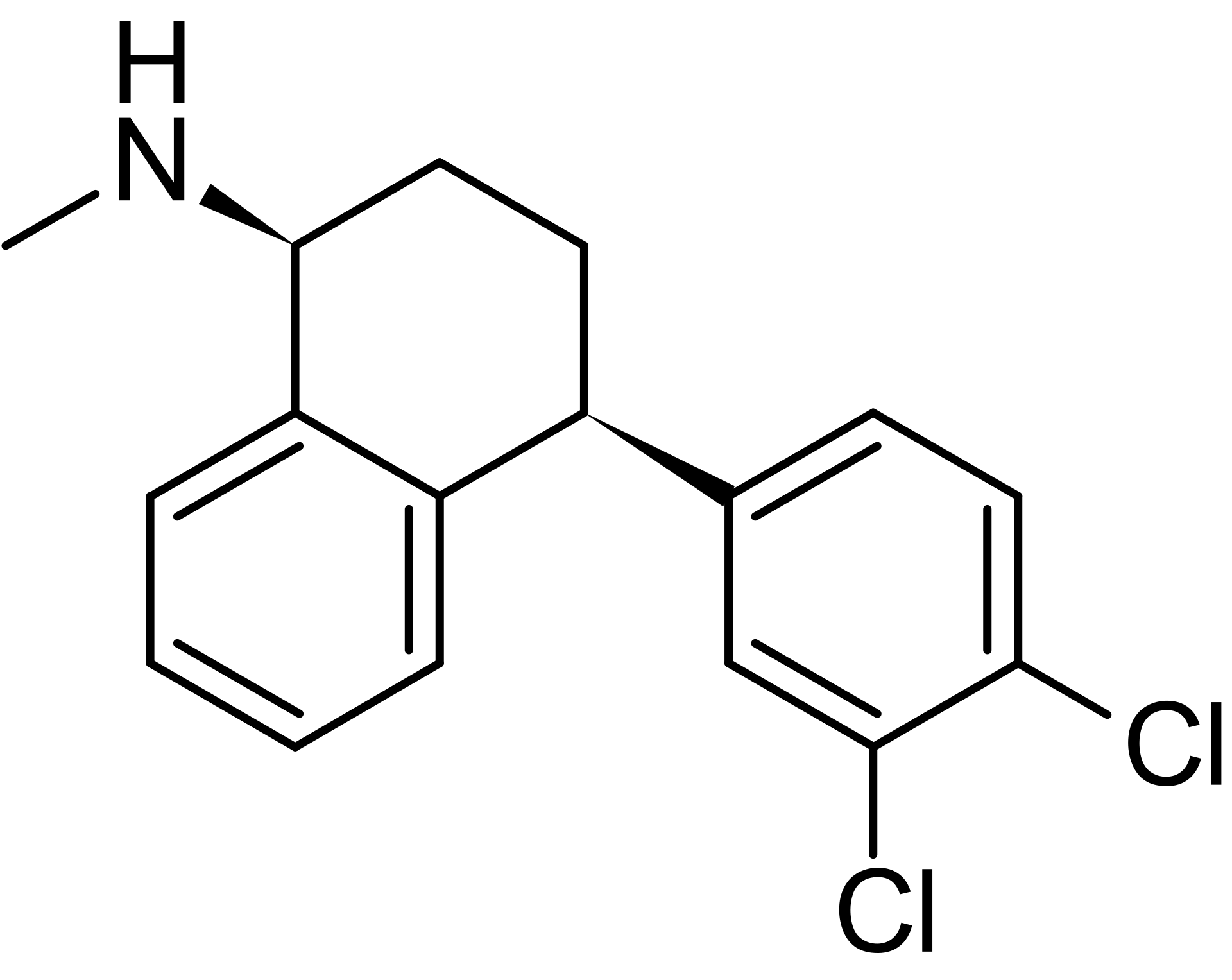
类别:选择性血清素再摄取抑制剂(SSRI)
NMPA核准适应症:重度抑郁障碍(MDD)、强迫症(OCD)、惊恐障碍、社交焦虑障碍、创伤后应激障碍(PTSD)、经前期烦躁障碍(PMDD)
厂家/规格/售价:
【原研】晖致制药(大连)有限公司 盐酸舍曲林片 50mg/14片 价格:72.49元
天津华津制药有限公司 盐酸舍曲林片 50mg/14片 价格:14.68元
江苏联环药业股份有限公司 盐酸舍曲林胶囊 50mg/14粒 价格:19.96元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点受体作用机制 | 主要抑制血清素转运体(SERT),提高突触间隙中血清素浓度,增强神经传导,轻微抑制多巴胺转运体,轻微拮抗Sigma-1受体 |
| 亲和力/选择性 | 对SERT的Ki值约为0.29 nM,显示出绝对上游的高亲和力和选择性 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 平均约26小时,范围13–45小时 |
| 起效时间 | 通常在服用后1–2周开始见效,4–6周达到最大疗效 |
| 起效周期 | 约4–6周达到稳态浓度和疗效峰值 |
| 首过效应 | 存在首过代谢,口服生物利用度约为44% |
| 吸收率 | 缓慢吸收,峰浓度出现时间为服用后4.5–8.4小时 |
| 活性代谢物 | 主要代谢产物为去甲舍曲林,活性较弱 |
| CYP参与信息 | 主要通过CYP3A4和CYP2C19代谢,CYP2B6次要辅助代谢,亦为CYP2D6的中度抑制剂 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 缓解速度 | 起效较快,部分患者在1–2周内症状改善 |
| 起效后临床表现 | 改善抑郁情绪、焦虑、强迫症状等,提升生活质量 |
| 不同精神症状适应性 | 对强迫症、焦虑障碍等疗效显著,亦适用于抑郁症等多种精神障碍 |
¶ 副作用
中枢神经类:头痛、失眠、嗜睡、焦虑、震颤等
性功能、体重:可能导致性欲减退、性功能障碍,体重变化
特殊副作用:可能引起胃肠道不适,如恶心、腹泻等;在青少年中,需警惕自杀念头的风险
¶ 优劣情况
明显优势:
对多种精神障碍有效,适应症广泛
起效较快,副作用相对可控
明显劣势:
性功能障碍、适应症广泛但并不完美适应
在青少年中需谨慎使用,监测自杀风险
因拮抗Sigma-1受体导致的明显认知损害(人话:降智、变笨、感觉懵乎乎的)
¶ 对于 MTF 群体的优劣情况
明显优势:真不行
明显劣势:代谢酶与多种HRT药物冲突!
用药注意
剂量:起始剂量通常为50mg/日,可根据疗效和耐受性调整,最大剂量不超过200mg/日
联用情况:避免与单胺氧化酶抑制剂(MAOI)联用,以防血清素综合征,ADHD患者慎用(因合成手段导致会多多少少影响一点多巴胺转运体,规避小概率事件)
雌二醇使用者慎用、CPA(色普龙)使用者慎用、比卡鲁胺使用者慎用
撤药换药须知:停药需逐渐减量,避免出现停药综合征
¶ 历史研发背景
舍曲林迭代自他美曲林(Tametraline,代号 CP-24,441),一款辉瑞公司早年放弃的化合物。而这背后的故事,源自一位传奇人物:Billie Kenneth Koe(1925年4月15日 – 2015年10月7日), 传奇天才医学生,华裔美国化学家,舍曲林的首席开发者。
1977年,在氟西汀问世五年后,Koe 博士在辉瑞重新关注起七年前公司开发、却被搁置的他美曲林。他美曲林是一种多巴胺再摄取抑制剂(DRI),也就是说它具备轻度精神兴奋效果,原理类似利他林,但作用远弱于后者的直接神经激活机制。由于涉及多巴胺系统的药物在药政审批中往往面临更严格审查,且成瘾风险较高,辉瑞最终选择放弃这条研发路线。
(他美曲林)
(舍曲林)
但七年后,Koe 博士与化学家 Willard Welch 决定探索新的方向——此时的学术界正开始关注 SSRI 类药物的巨大潜力。他们意识到,为了绕开已有药物的专利壁垒,他们必须设计一款具有原创结构的 SSRI。他美曲林,作为一款已有合成基础的 DRI,也因此成为了改造对象。
将一个多巴胺抑制剂改造为选择性血清素再摄取抑制剂,从理论上说流程简单——删去多巴胺段,替换为血清素作用段。但在化学操作层面,这仍是暴力而复杂的工程。Koe 博士采用了 Alexander Shulgin 舒尔金博士常用化学爆改的手法:在苯环的3位和4位引入两个氯分子,再在顶端结构上做出关键微调,再筛选为1S/4S异构体。所以舍曲林曾经也被叫做3,4-二氯-他美曲林。
由于这种“非传统路径”的构造方式,舍曲林的化学结构在 SSRI 家族中显得极为独特——不像氟西汀那样的左右端对称,也不像帕罗西汀致密。
舍曲林经过异构体筛选,最终确定最优构型为 1S,4S 异构体,并进入临床开发。舍曲林于 1991年完成三期临床并获批上市,迅速在全球铺开销售。时至今日,舍曲林与艾司西酞普兰一道,成为全球开具量最大的抗抑郁药之一。在中国,它是临床最常用的 SSRI,尤其在青少年人群中具有极高开具率。
Koe 博士于 2015 年离世,享年 90 岁。他曾在 2008 年荣获辉瑞的 霍华德·沃勒姆成就奖,并在获奖感言中说道:
“我和我的同事们都感到既敬畏又欣慰,因为我们的努力最终研制出了一种能够帮助病人的世界级药物。不止一次,陌生人、熟人和服用舍曲林后受益的朋友都来亲自感谢我为这种抗抑郁药的发现所做出的贡献。我在舍曲林的发现过程中所扮演的角色取决于许多因素的共同作用。个人必备条件包括扎实的技术背景、学习和适应的意愿、准备充分的头脑以及毅力。拥有和蔼可亲的同事和通情达理的老板也很重要。所有药物研发工作中的一个关键因素仍然是运气!我很幸运地参与了制药行业的黄金时代,这给了我一个独特的机会,让我能够创造性地思考,并在化学专业之外的技术领域从事科学研究。”
Koe 是一个真正的传奇人物。童年时期与家人颠沛流离,靠洗碗维持生计,后来考入林肯高中,并凭借全额奖学金进入里德学院,之后取得华盛顿大学硕士和加州理工学院博士学位。他最初在辉瑞从事抗生素开发,最终却为世界带来了第二款 SSRI,也是划时代意义的抗抑郁药——舍曲林。
中国获批和医保时间轴:
NMPA批准时间晚(2023),仿制药早于原研流入中国市场,具体时间已不得而知
专利期时间:
2006年6月30日,已过期,有多种仿制药上市
¶ 主观评分: 6.5/10(常规群体) 4.5/10(HRT群体)
¶ 主观评价
其实如果单从中国医学和用药方面来评论的话,舍曲林无法达到6.5分的评价,最多6分,这额外的0.5是因为,Koe博士是影响我很大的人,是一个伟大的、不甘于现状而逆流而上的传奇,或许舍曲林的开发中,Koe博士可能受到了舒尔金的指点,但筛选异构体、基于DRI开发依然是一个人类史上至今无法复刻的举动,这款药也在很长一段时间带给很多抑郁症、焦虑障碍等患者一道光。
但从2025年的角度来看,舍曲林也存在许多问题,第一:舍曲林的适用面太广了,基本算是SSRI里适用面最广的药物,这无疑是好事,但是这也带来了一个隐性问题:在医疗资源紧张的中国,尤其是精神科医生的缺乏,再加上精神科用药本来就存在“一药多病,一病多药”,舍曲林就成为了最适合“流水线”式开具的药物,许多具有特殊性的抑郁症、焦虑障碍患者最适合的用药并不是舍曲林,但依然会被开具到舍曲林。对于临床医生来说,这也是有一定道理的,舍曲林是他们最为了解、临床案例最多的药物,所以“相对最稳定最安全”也成为了舍曲林被开具的原因。
从化学角度来说,舍曲林因为DRI迭代的缘故,具备了多巴胺的微弱驱动力以及对Sigma-1受体的拮抗,成为了它适用面广泛的原因,但首先,Sigma1受体拮抗就是导致舍曲林使用者“变笨变呆变人机”的主要成因!也是一些例如ADHD等疾病使用时风险增加的成因,且舍曲林的吸收率和首过效应波动太大了,这就会导致十个人吃舍曲林会有十种评价。舍曲林的性功能损害相对算低,但是还是高于艾司西酞普兰许多。而针对MTF群体而言,舍曲林在部分人群中被证实升高催乳素,会导致HRT人群发生极罕见的乳腺结节风险,而更重要的是,舍曲林有部分(10%~15%)依靠CYP3A4代谢,而HRT常见的药物里,补佳乐、CPA、比卡鲁胺全部都是CYP3A4为主代谢,联用会发生撞车,导致两边的血药浓度上升或下降,也就是两边的效果都不稳定,尽管没有专门的资料去研究血药浓度波动大小且舍曲林的CYP3A4占比并不高,有CYP2C19/CYP2B6/CYP2D6分摊代谢导致CYP3A4只占到10%~15%,但在市场上这么多SSRI药物可选的情况下,安全起见将舍曲林替换掉是最好的选择。因此,对于HRT群体来说,强烈不推荐使用舍曲林,由于临床医生很少会考虑到HRT因素,所以如果作为HRT治疗中的患者,你被开到了舍曲林,请去和你的医生说明情况并更换至不依赖CYP3A4代谢的抗抑郁药。(依赖CYP3A4的抗抑郁药物有:舍曲林、艾司西酞普兰、曲唑酮、伏硫西汀)
¶ 西酞普兰(Citalopram) / 艾司西酞普兰(Escitalopram)Lu-10-134-D / Lu-26-054-0【HRT群体不作为首要推荐用药,原因参考下方评价】
¶ 基本信息
药物国际通用名/商品名:西酞普兰 / 喜太乐(Celexa)、艾司西酞普兰 / 来士普(Lexapro)
化学分子式:C20H21FN2O(两者相同,但艾司西酞普兰为S-对映体)
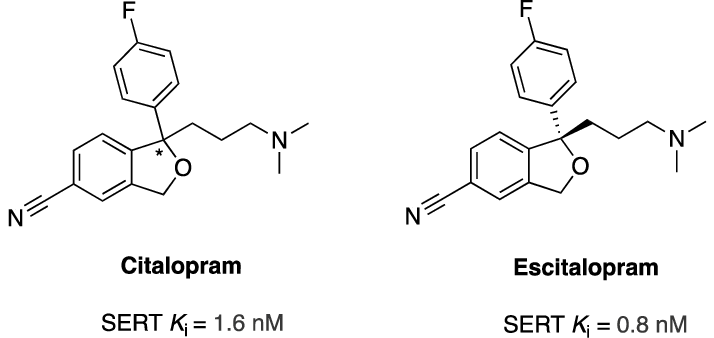
类别:选择性血清素再摄取抑制剂(SSRI)
NMPA核准适应症:重度抑郁障碍(MDD);艾司西酞普兰另适用于广泛性焦虑障碍(GAD)
厂家/规格/售价:
【原研】H. Lundbeck A/S 氢溴酸西酞普兰片 20mg/14片 价格:108.47元
四川科伦药业股份有限公司 氢溴酸西酞普兰片 20mg/14片 价格:32.84元
【原研】H.Lundbeck A/S 草酸艾司西酞普兰片 10mg/7片 价格:77.08元
四川科伦药业股份有限公司 草酸艾司西酞普兰片 10mg/14片 价格:30.35元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 主要受体作用机制 | 选择性阻断突触前膜的血清素转运体(SERT),提升突触间5-HT浓度,改善情绪状态与焦虑反应 |
| 亲和力/选择性 | 西酞普兰为R+S消旋混合体,R-异构体无效甚至产生拮抗,艾司西酞普兰为S-对映体,亲和力高、作用更纯净、剂量更低即达效。Ki约0.29 nM,为SSRI中较强者之一 |
*艾司西酞普兰是公认的最“干净”的SSRI,对其他受体亲和力几乎全部为0,它只专注于血清素转运体,这也就导致:
更专一 → 更耐受:艾司西酞普兰因无组胺/胆碱/多巴胺/去甲肾上腺素受体活性,因此不太会引起嗜睡、体重增长、口干、便秘、性功能严重障碍等问题,耐受性普遍优于其他SSRI;
焦虑改善来源于SERT特异性:不像氟伏沙明依赖Sigma1,或氟西汀通过5-HT2C,艾司西酞普兰的焦虑缓解可能纯粹源于对SERT的高亲和占位,降低5-HT负反馈抑制,增加前额叶投射;
但其焦虑维度以GAD为主,非Sigma1型“抗恐惧”效应较弱,不太适合社交恐惧、创伤后焦虑等情境。
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 平均约27–32小时,范围约20–45小时,艾司西酞普兰略短于西酞普兰 |
| 起效时间 | 通常在服用后1–2周开始见效,4–6周达到最大疗效 |
| 起效周期 | 稳定状态与疗效峰值通常出现在连续服用4–6周后 |
| 首过效应 | 存在首过代谢,口服生物利用度约为80% |
| 吸收率 | 缓慢吸收,峰浓度出现在服药后约4.5–5小时之间 |
| 活性代谢物 | 艾司西酞普兰的主要代谢产物为去甲艾司西酞普兰,活性较弱 |
| CYP参与信息 | 主要通过CYP2C19与CYP3A4代谢,辅以CYP2D6,CYP3A4依赖度较高(≈30%),MTF群体联合HRT时需注意酶竞争交互影响 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 缓解速度 | 艾司西酞普兰通常起效更快,部分患者1周即出现焦虑缓解; |
| 起效后表现 | 情绪稳定、睡眠恢复、焦虑减轻、兴趣增强、认知活力提升 |
| 精神适应性 | 艾司西酞普兰对广泛性焦虑障碍适配度极高,对“认知耗竭型”“高警觉型”抑郁亦有效。西酞普兰适用于情绪淡漠型/广谱型MDD患者,疗效稳定但渐进慢热。 |
¶ 副作用
两者常见的偶然副作用包括:失眠、乏力、胃肠不适、性欲减退、性高潮延迟、射精障碍等,属于典型SSRI副作用谱。但在所有SSRI中艾司西酞普兰的副作用出现率最低,是目前市场上最稳定的SSRI之一。
西酞普兰在剂量 >40mg/d 时QT延长风险显著,65岁以上患者推荐剂量不超过20mg。艾司西酞普兰QT风险略低,但仍建议心电监测。部分患者可能在初期出现激越、坐立不安,通常可随时间缓解。
¶ 优劣分析
西酞普兰优势在于价格低廉、血药浓度稳定、QT管理得当时适配人群广泛;缺点在于活性混杂,R异构体影响效力,焦虑改善效果较弱。
艾司西酞普兰是西酞普兰的“提纯版”,纯S-构型使得剂量更低、起效更快、焦虑改善更显著,副作用整体较轻。
雌二醇使用者慎用、CPA(色普龙)使用者慎用、比卡鲁胺使用者慎用
艾司西酞普兰依赖CYP3A4/CYP2C19代谢路径,在联用雌激素类HRT药物(如雌二醇、比卡鲁胺)时存在酶竞争风险,需个体化调整,必要时避开共用代谢通道的药物。
¶ 对MTF群体的适配性
艾司西酞普兰在焦虑缓解方面表现优秀,原本为MTF群体(尤其是HRT初期激素波动明显、性别焦虑较重者)推荐SSRI之一。然而,其CYP3A4依赖度高,与多种HRT药物(雌二醇、比卡鲁胺、螺内酯)代谢通路发生重叠。此代谢冲突可能造成两者血药浓度异常,带来激素不稳定或药效增强/削弱等问题。
因此,对于已使用或计划使用雌激素凝胶、比卡鲁胺的MTF患者,建议在医生指导下评估艾司西酞普兰的CYP交互风险,可优先考虑代谢更明确、交互风险更低的SSRI(如氟伏沙明、帕罗西汀)。
西酞普兰则不推荐用于焦虑主导的MTF患者,因其对焦虑症状支持力弱,激越风险高,且QT延长限制较大。
¶ 历史与研发背景
西酞普兰家族在抗抑郁药历史上起到的一大作用就是:它证明了去甲肾上腺素不是导致抑郁症的关键因素,而是血清素,而这一切的挖掘者,通过一个小小的思路转换,就做出了这款撼动医学界的纯粹的SSRI。
Klaus Bøgesø,西酞普兰的发明人,以下简称克劳斯。
整个西酞普兰的追溯历史,其实已经接近70年。
艾司西酞普兰迭代自西酞普兰,西酞普兰迭代自塔罗普兰(Talopram , Lu-3010),塔罗普兰则迭代自三环类抗抑郁药美利曲辛,美利曲辛则是阿米替林的升级版。阿米替林是美国默克公司于1957开发的专利药品,灵北制药厂在1963年开发出了基于阿米替林的速效升级版化合物-美利曲辛,并取得了专利。
而1971年,灵北制药厂雇佣克劳斯,希望他能在灵北开发的美利曲辛基础上,提纯出更针对性靶向抗抑郁的药,1971年时血清素假说还没有被广泛采用,灵北的思路是开发一款去甲肾上腺素再摄取抑制剂(NRI),在当时的年代背景下,三环类抗抑郁药起效的具体原因没有人知道,三环类抗抑郁药就像是一把霰弹枪,它把所有的靶子都打到了,其中的某个靶子起了效果能抗抑郁,而灵北当时认为起效果的靶子,是去甲肾上腺素。于是克劳斯将美利曲辛进行提纯,制作出了两款衍生物:塔罗普兰和塔苏普兰,它们很符合灵北制药厂的需求,但在临床试验中险些出现事故:实验发现,受试者变得更加活泼、好动、警觉,但并没有改善抑郁,甚至加重了受试者的抑郁,灵北制药厂担心后续的风险,于是停止了塔罗普兰和塔苏普兰的临床试验并废弃了此项目。
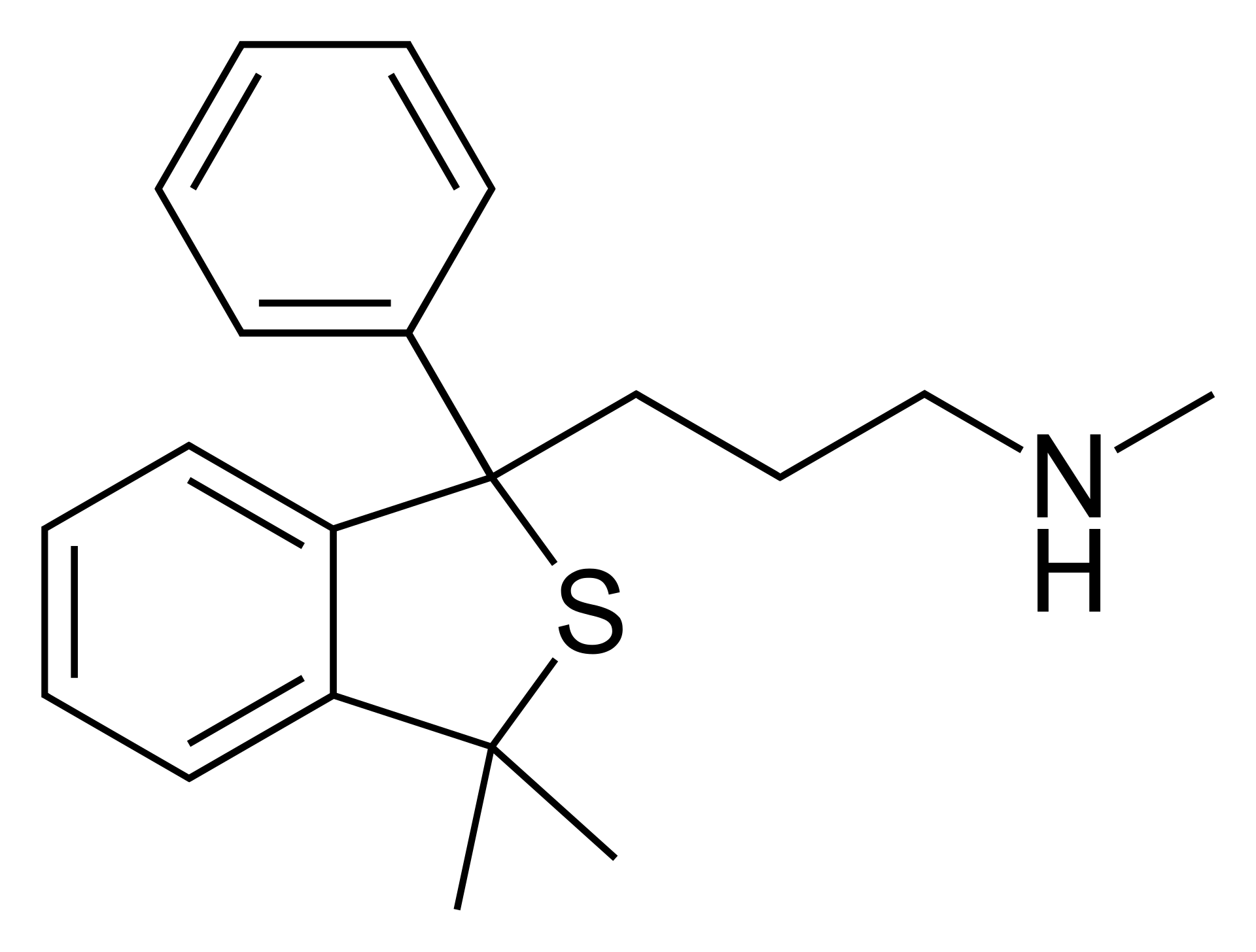
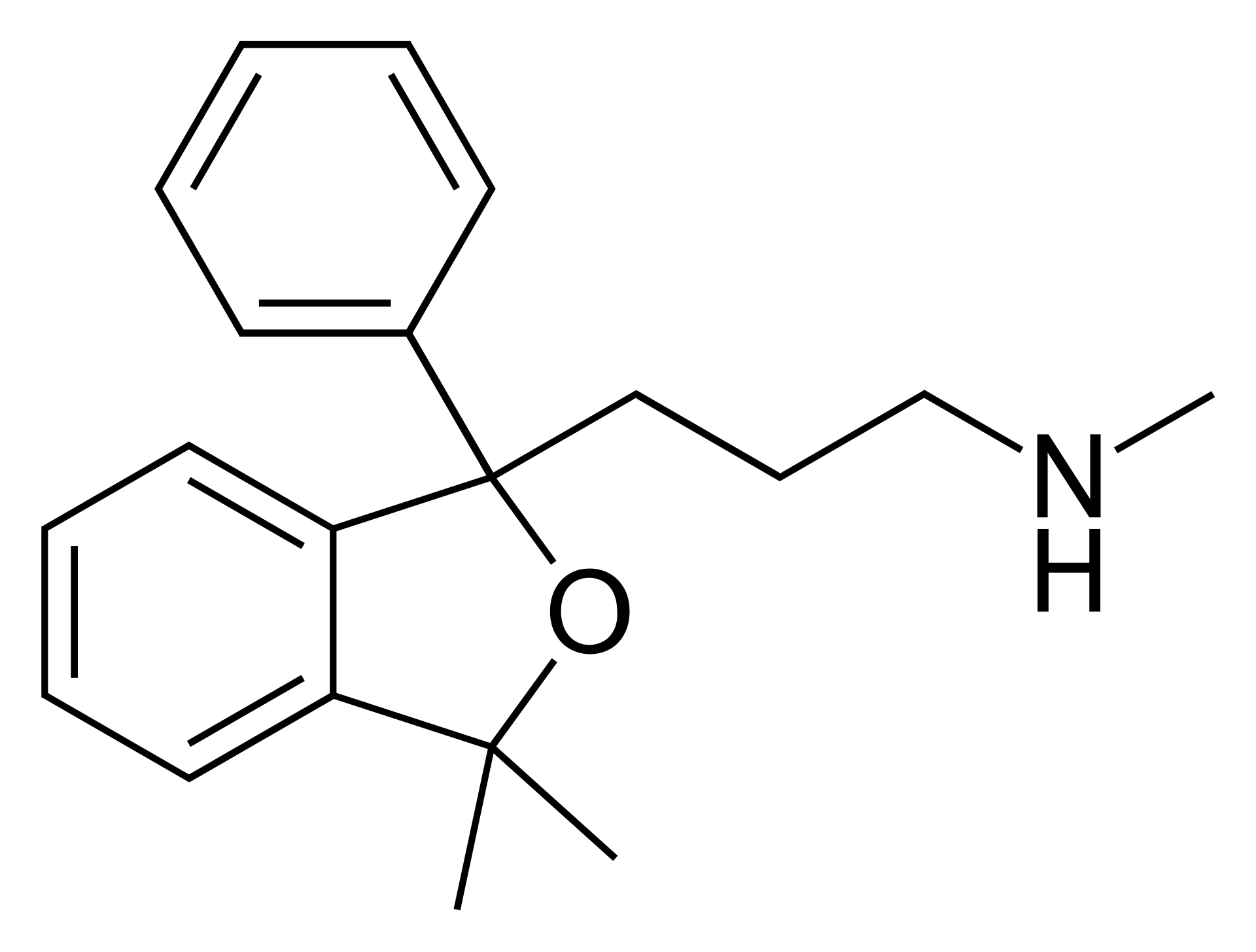
但克劳斯并未放弃它们,他反复思考临床实验失败的核心因素,塔罗普兰和塔苏普兰的开发需求是:必须是去甲肾上腺素再摄取抑制剂,那么有没有一种可能,灵北制药厂的NE致郁理论是错误的?通过相当长时间的科研,他推翻了NE致郁的假说,认为5HT(血清素)才是导致抑郁的关键,那么如果只针对性的增强脑内的血清素水平,是否会将死去的塔罗普兰/塔苏普兰复活?
于是克劳斯选择塔罗普兰作为基础,同样用了美国化学家Alexander Shulgin 舒尔金大佬 的手法(有趣的是,舍曲林的开发在苯环3、4位插了氯,动苯环34位是舒尔金常用的gimmick,很有可能Koe博士和克劳斯都受到了舒尔金大佬的指点。),在右苯环4位插上氟,左苯环5位加上氰基,再对甲基进行简易加减,在1989年,来自已经死去的塔罗普兰的新生命-西酞普兰横空出世。
由于西酞普兰是从非常纯粹干净的去甲肾上腺素再摄取抑制剂改装而来,它也继承了【干净】的特性,经过测试后发现西酞普兰对血清素的选择性在当时达到了全部抗抑郁药中的最高水平,约为舍曲林的2.5倍,氟西汀的110倍。(此测试的内容是该化合物中对血清素转运体(SERT)和去甲肾上腺素转运体(NET)的选择性差值,即Ki_NET / Ki_SERT,西酞普兰在当时测试出来的结果是约为3333倍,地表最强,舍曲林为1390倍。而到如今,西酞普兰的左旋体-艾司西酞普兰接管了【地球第一纯度】的皇冠,选择性比例高达7100倍。)西酞普兰的专利在2003年过期,为了继续在这款药上盈利,灵北制药将西酞普兰进行手性分离后得到的左旋体-艾司西酞普兰注册了专利,左旋体是更更更纯粹的西酞普兰,效果是右旋体的27倍。2012年12月,克劳斯亲自告诉了全世界40年前他如何发明的西酞普兰,他是一个严谨而又坚定的伟大化学家,正是他对塔罗普兰和塔苏普兰失败的仔细反思和推论后,才有了西酞普兰,而塔罗普兰、塔苏普兰、西酞普兰、艾司西酞普兰全部都是克劳斯手下的产物,因此提到西酞普兰家族,就一定会提到克劳斯。如今,艾司西酞普兰的专利也已经过期,仿制药将艾司西酞普兰降到了很低的价格,再加上国内挂网价格的谈判和医保的介入,使艾司西酞普兰成为了国内性价比最高的抗抑郁药之一。
¶ 西酞普兰家族树时间轴
| 年代 | 事件 |
|---|---|
| 1957年 | 默克公司:阿米替林(Amitriptyline)问世 |
| 1960s | 灵北制药:美利曲辛(Meritriptine)问世 |
| 1971年 | 克劳斯研发塔罗普兰与塔苏普兰,项目因不良反应停止 |
| 1989年 | 克劳斯发明西酞普兰(Citalopram) |
| 2001年 | 艾司西酞普兰(Escitalopram)专利注册 |
| 2003年 | 西酞普兰专利到期,全球仿制药兴起 |
| 2012年 | 克劳斯公开西酞普兰发明全过程 |
西酞普兰家族不仅是药物化学领域的里程碑,也代表了抗抑郁药从“多胺提升”到“精准血清素调控”的范式转变。提及这一家族,就必须致敬 克劳斯·伯格索——正是他在失败中坚持反思与突破,才有了今天全球数千万抑郁患者的福音。
¶ 主观评分:8.5/10 (艾司西酞普兰/常规群体) 7/10 (艾司西酞普兰/HRT群体) 6/10(西酞普兰/常规群体) 5/10(西酞普兰/HRT群体)
¶ 主观评价:
艾司西酞普兰和舍曲林,现在抗抑郁药市场的海尔兄弟,舒克与贝塔,但艾司西酞普兰的风评比舍曲林好一些,主要原因归功于艾司西酞普兰实在是太™干净了,这东西能干到7100倍的NET/SERT Ki选择对比值,地表最强,意味着艾司西酞普兰是基本绝对意义上的只碰血清素的抗抑郁药了,至于舍曲林,舍曲林嗷,你小子拮抗Sigma1受体,让人变笨,你兄弟艾司西酞普兰纯的一批,比你好多了!
但是注意,我给常规群体评分8.5的主要原因是:1.艾司西酞普兰的货量大,基本上每个医院都有,是你针对性换药的最好选择。 2.艾司西酞普兰是国内医生们了解度仅次于舍曲林的药物,开这个药给患者,医生够放心。
不过同样,西酞普兰的6分也并不意味着一无是处,干净代表着对症下药的精准,混悬体代表着能治疗的精神障碍要多一丢丢。艾司西酞普兰只适合常规的抑郁症、重度抑郁症和广泛性焦虑障碍,一些特殊成因的抑郁症和双相,是需要血清素以外的受体变动的。
作为SSRI,艾司西酞普兰做到了最纯,但我个人(仅限个人看法!)对5HT致郁学说抱有深刻怀疑态度,虽然我也并不是很相信如今抗抑郁领域正在研究的NMDA致郁学,我个人更倾向于老保派,也就是SND全部变动,只是需要更合适的比例。(加州火箭燃料【米氮平联用文拉法辛】能够那么显著的看到效果 就是因为动的受体足够多且比例合理)但我毕竟不是专业的科研人员,我的观点代表不了什么,只是根据如今SSRI的高概率受体不应答、不起效而做出的怀疑罢了。
P.S. 艾司西酞普兰和舍曲林还真是俩兄弟,一个迭代自NRI,一个迭代自DRI,不同的道路通向了同一个城市,顶峰相见。
感谢每一位抗抑郁药领域的科研人员为精神疾病患者做出的杰出贡献。
P.S.2. 艾司西酞普兰的药代动力学牵扯到了30%~35%左右的CYP3A4代谢,这会影响HRT药物的浓度平衡也会影响它自身,虽然没有专项研究证明波动大小且艾司西酞普兰依赖多酶分摊代谢,有CYP2C19/CYP2D6来分摊,但CYP3A4的纸面占比数据无可置疑的还是来到了30%~35%,为了安全起见,不建议HRT中的MTF群体选择艾司西酞普兰,严谨用药,生命安全第一位。
¶ 帕罗西汀(Paroxetine)GSK‐7051/ [FG-7051] 【HRT群体不作为首要推荐用药,原因参考下方评价】
¶ 基本信息
药物国际通用名/商品名:帕罗西汀 / 赛乐特(Paxil)
化学分子式:C₁₉H₂₀FNO₃
类别:选择性血清素再摄取抑制剂(SSRI)
NMPA 核准适应症:重度抑郁障碍(MDD)、强迫症(OCD)、惊恐障碍(PD)、创伤后应激障碍(PTSD)、社交焦虑障碍(SAD)
厂家/规格/售价:
石家庄龙泽制药股份有限公司 盐酸帕罗西汀片 20mg/28片 价格:18.80元
【原研】GlaxoSmithKline 葛兰素史克(天津)有限公司 盐酸帕罗西汀片 20mg/10片 价格:59.29 元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点/受体作用机制 | 高效抑制突触前血清素转运体(SERT),增强 5-HT 在突触间的持续作用 |
| 亲和力/选择性 | 对 SERT 具有亚纳摩尔级亲和力;对去甲肾上腺素转运体(NET)和多巴胺转运体(DAT)作用有但极小 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 约 21–24 小时 |
| 达峰时间(Tmax) | 5–9 小时 |
| 稳态时间 | 连续用药约 7 天可达稳态 |
| 生物利用度/吸收 | 约 50%,食物影响轻微 |
| 代谢/清除 | 主要经肝脏 CYP2D6(次CYP3A4、CYP2B6)代谢;少量原药经肾排泄,稳态后CYP3A4代谢比例≈35%。 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 起效速度 | 初效需 2–4 周,4–8 周疗效显著 |
| 起效后表现 | 情绪稳定、焦虑缓解、睡眠与食欲逐步恢复 |
| 精神症状适应性 | 对强迫症、惊恐发作及创伤后应激障碍疗效确切 |
¶ 副作用
中枢神经系统:头痛、失眠或嗜睡、激越、焦虑短暂加重、做梦频率增加、做噩梦、梦魇
性功能与体重:性欲减退、勃起/射精障碍;部分患者体重显著变化
特殊警示:偶见躁狂转换、QT 间期延长、癫痫发作风险
青少年及双相患者使用时可能出现自杀念头或躁狂倾向,需严密监测 (SSRI通用)
¶ 优劣情况
优势:对 OCD、GAD、PTSD 等多种适应症覆盖面广;起效相对其他 SSRI 稳定
劣势:性副作用突出;停药可出现严重撤药综合征;超绝TOP1抗胆碱能
¶ 对于 MTF 群体的优劣
明显优势:抗焦虑效果较快显现,早期激动性较低
明显劣势:性功能副作用加重,可能影响性别认同体验
虽然单剂量时帕罗西汀主要经 CYP2D6 代谢(≈98%),但在稳态条件下(连续给药后 CYP2D6 通路部分饱和),CYP3A4 对其清除的贡献可上升至约 35%;CYP2D6 的比例则从 ≈98% 降至 ≈44%。
因此,在 HRT 患者同时给药时,帕罗西汀和雌激素制剂会争用同一 CYP3A4 通路,可能导致:
-帕罗西汀血药浓度波动,疗效及副作用不稳;
-雌激素药物代谢减慢,雌激素水平升高,增加血栓、乳腺增生风险。
¶ 用药注意
剂量:成人起始 20 mg/日,餐中或餐后服用;如耐受良好可逐步增至最大 50 mg/日
联用:慎与苯二氮卓类、MAO 抑制剂、HRT药物及 QT 延长药物合用
撤药建议:逐步减量至少 2–4 周,避免突然停药诱发撤药症状
¶ 历史与研发背景
帕罗西汀是整个SSRI家族中最为神秘的一个成员。与其他药物相比,它的诞生历程似乎更像一个迷雾重重的故事,网络与公开文献中极少提及它的起源,仿佛开发者们刻意隐藏了某些秘密。帕罗西汀的起点可能远比我们想象的更加戏剧化和复杂,而这一切的关键,或许就隐藏在20世纪早期广泛使用、后来臭名昭著的可卡因之中。
在20世纪中期,科研人员发现,可卡因作为一种强效的多巴胺、去甲肾上腺素、血清素三重再摄取抑制剂(SND激动剂),具有显著的情绪提升效果,但同时也伴随极强的成瘾性和严重的毒副作用。由此,科学家们萌生了一个大胆而关键的想法:是否能够从可卡因的结构中提取出一种仅专注于血清素系统(5-HT)的药物,以此避免多巴胺通路的成瘾性与毒性副作用?
在1970年代,随着氟西汀的成功问世,选择性血清素再摄取抑制剂(SSRI)概念初步获得医学界的肯定,全球多家药企迅速投入SSRI的研发热潮之中。此时丹麦的一家制药公司费罗森(Ferrosan A/S)也秘密加入了这场竞赛,试图从更深远的历史背景中寻找灵感。
他们的研究路线似乎与其他公司截然不同,费罗森团队从莨菪烷类天然产物的结构入手,CPCA 诺卡因,以及前体更简单的起源 槟榔碱,这些化合物最初都与可卡因具有结构上的血缘关系。他们进行结构骨架的简化与优化,将复杂的莨菪烷双环骨架转化为更为简单而稳定的哌啶骨架,并最终得到了一种名为FG4963(非莫西汀)的化合物。尽管FG4963最初在体外测试中展现了优异的选择性血清素再摄取抑制能力,但却因为其体内代谢周期过长、药代动力学表现不佳等原因而被迫放弃。
-cpca.svg.png)
费罗森并未停下脚步,他们又在FG4963的基础上进行了进一步的结构调整:在哌啶骨架上引入了对氟苯基基团以提高药物对血清素转运蛋白(SERT)的亲和力;同时加入了1,3-苯并二氧杂环这一独特的结构,以增强药物的代谢稳定性和选择性。这种精妙的结构重排与修饰,让他们最终获得了一个全新的化合物——FG7051,这就是后来的帕罗西汀。
但与此同时,费罗森团队还尝试了一种名为FG7080的化合物,它实际上是FG7051帕罗西汀与FG4963非莫西汀的混合物,尽管它们最初被寄予厚望,最终却因多种药理矛盾问题无法进入临床应用。
1980年,费罗森公司向英国的必成(Beecham)公司转让了FG7051的独家开发、生产及销售权,而费罗森本身则在1986年被丹麦Novo Industri公司收购,并于1989年与Nordisk Gentofte合并成为如今享誉全球的诺和诺德(Novo Nordisk)。
而另一边,接手帕罗西汀开发的必成公司(Beecham)则经历了戏剧化的转变:1989年与SmithKline Beckman公司合并,成立了SmithKline Beecham公司。经过大量严谨的临床研究与药理实验后,他们终于在1992年成功将帕罗西汀(商品名Paxil/赛乐特)推向市场。但巧合的是,同年帕罗西汀的初始专利即告到期,专利权的风雨飘摇也预示了它此后坎坷的发展历程。
2000年,SmithKline Beecham与Glaxo Wellcome合并,最终诞生了如今全球知名的药企巨头——葛兰素史克(GSK)。至此,帕罗西汀也由FG7051正式更名为GSK7051。
纵观帕罗西汀的整个历史迭代,就像一个流浪汉一般四处辗转,历经重重困难与不确定性。它并不像氟西汀、西酞普兰那样光明正大地出现在历史舞台之上,而是经历了隐秘而艰辛的道路。然而,也正是这些复杂而神秘的迭代历程,造就了帕罗西汀独特的药理特性与稳态代谢变动特征。
如今的帕罗西汀,凭借其明确的临床疗效、多种适应症(如抑郁症、强迫症、焦虑障碍)以及复杂却稳定的药代动力学,已经成为全球范围内重要的抗抑郁药物之一。尽管其起源故事充满了迷雾与猜测,但不可否认的是,帕罗西汀在抗抑郁药物的历史长河中拥有其不可替代的位置,它以特殊的药物迭代史,见证了现代药物化学从“复杂多靶点作用”向“精准血清素调控”的转变之路。
因此,提起帕罗西汀,我们不仅要理解它的复杂迭代历程,更要敬佩那些隐藏在幕后的科研人员——正是他们在迷雾中摸索与坚持,才让全球无数抑郁症患者获得了疗愈的希望。
¶ 主观评分: 7/10(常规群体) 5/10(HRT群体)
¶ 主观评价
在SSRI的家族中,帕罗西汀是一款极具争议且特殊的存在。单从纸面数据来看,这款药确实惊艳无比:它对血清素转运体(SERT)的Ki值低到令人震惊的中位数0.07–0.13 nM,无可置疑是SSRI家族中最强的血清素亲和力药物。然而,我个人对帕罗西汀并没有太好的评价,反而带着更多的批判与谨慎。
首先,让我们谈谈MTF群体与帕罗西汀之间复杂且微妙的关系。从表面数据看,帕罗西汀的CYP3A4代谢仅占初始给药的2%,这种程度的参与通常不会被过度关注,但问题恰恰出在长期使用后的稳态阶段。在稳态条件下,CYP2D6通路逐渐饱和,帕罗西汀被迫更多地依赖CYP3A4通路进行代谢,比例甚至高达35%。而我们知道,绝大多数MTF群体使用的HRT药物,例如补佳乐、比卡鲁胺、CPA等,都是以CYP3A4作为主要代谢通路。这意味着长期使用帕罗西汀将不可避免地与这些药物发生酶竞争冲突,直接导致双方血药浓度的不稳定。这种冲突风险尽管尚未有大量临床数据具体呈现波动范围,但出于安全起见,我强烈不推荐MTF群体选择帕罗西汀作为抗抑郁药物,如果被开具到,应主动向医生说明情况,更换为代谢通路更安全的抗抑郁药物。
其次,作为长期服用过帕罗西汀的人,我必须强调一下它最大的困扰之一:噩梦甚至是严重的诡异恐怖梦魇问题。这并非个例,而是广泛被患者抱怨的常规现象。究其根本原因,帕罗西汀在所有SSRI中拥有最强烈的抗胆碱能作用,远远超过舍曲林、西酞普兰甚至氟西汀。胆碱能系统在睡眠过程中主要负责调控快速眼动睡眠(REM)期的启动与维持,抗胆碱作用严重时会扰乱REM睡眠的结构,导致REM期变得破碎、不连贯,甚至严重紊乱。这种紊乱状态往往表现为强烈而怪异的梦境体验,负面梦境的发生率大幅提高,甚至变为挥之不去的梦魇。
此外,帕罗西汀尽管以强效的SERT抑制著称,但实际上,它还带有一定的NET(去甲肾上腺素转运体)抑制作用,尽管这个作用非常轻微(且是需要达到≥40mg),但已足以让帕罗西汀沾染上一丝SNRI的特质。去甲肾上腺素在睡眠时本应处于低活跃状态,过高的NE水平却会明显增加觉醒频率、夜间清醒次数,甚至加剧交感神经兴奋,临床研究已显示,这种交感神经过度激活与梦魇存在明确关联。因此,对于存在睡眠问题或对梦境异常敏感的患者,帕罗西汀绝非最佳选择。
总的来说,尽管帕罗西汀在血清素选择性上的极端表现理论上能给予某些重度抑郁症患者精准而强大的帮助,但临床现实并非如此单纯。抗胆碱副作用所带来的梦魇问题,稳态下CYP3A4代谢比例暴涨所引发的药物相互作用风险,都使得帕罗西汀成为一款需要非常谨慎选择的药物。事实上,绝大多数患者并不需要如此极端的血清素再摄取抑制效果,而是更需要药效、副作用与安全性之间更合理的平衡。因此,与其单纯追求“最强SSRI”,许多情况下,或许更为有效而平衡的治疗组合(比如“加州火箭燃料”文拉法辛联用米氮平)反而是更好的选择。
谨慎用药,生命安全与治疗体验永远应放在首位。
感谢每一位在抗抑郁药领域辛勤工作的科研人员,正是你们的贡献,才使我们得以安全地对抗抑郁症。
P.S. 如果你是正在HRT治疗中的MTF患者,请务必与医生详细沟通代谢酶冲突问题,帕罗西汀并非安全的选择,慎之又慎。
¶ 伏硫西汀(Vortioxetine)LU-AA21004
基本信息
药物国际通用名/商品名:伏硫西汀 / 心达悦(Brintellix / Trintellix)
化学分子式:C₁₈H₂₂N₂S
类别:多模态血清素再摄取抑制剂(MSRI)多模态抗抑郁药 (SMS)(Serotonin Modulator and Stimulator)
*伏硫西汀在药理上兼具高度SERT抑制活性与多种5-HT受体亚型的激动/拮抗作用,属于一种功能上的SSRI,并常被归类于‘新型SSRI’或‘多模态SSRI’,但其药理学机制已超越传统SSRI范畴
NMPA核准适应症:重度抑郁障碍(MDD)
厂家/规格/售价:
成都康弘药业集团股份有限公司 氢溴酸伏硫西汀片 10mg/14片 价格:25.01元
【原研】灵北制药(H. Lundbeck) 氢溴酸伏硫西汀片 10mg/14片 价格:497.99元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点受体作用机制 | 血清素(5-HT)再摄取抑制剂(SRI),同时激动5-HT₁A受体、部分激动5-HT₁B受体,拮抗5-HT₃、5-HT₇及5-HT₁D受体 |
| 亲和力/选择性 | 高度多模态作用,5-HT转运体抑制活性极高,同时对5-HT₃拮抗强,其他5-HT亚型受体亲和中等,去甲肾上腺素与多巴胺转运体亲和极低,明显区别于传统单纯SSRI类 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 约66小时(长期稳态) |
| 达峰时间(Tmax) | 7–11小时 |
| 稳态时间 | 约14天达到稳态 |
| 生物利用度/吸收 | 生物利用度高达75%,不受食物明显影响 |
| 代谢途径 | 主要由CYP2D6代谢为惰性代谢物,CYP3A4、CYP2C9、CYP2C19次要参与 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 缓解速度 | 起效较快,通常1–2周初效,4–6周显著疗效 |
| 起效后表现 | 情绪改善、认知提升、焦虑缓解、日常功能恢复良好 |
| 精神症状适应性 | 对伴认知功能减退、注意力问题、焦虑合并抑郁的患者尤为适合 |
副作用
中枢神经系统:恶心、头痛、头晕、偶见失眠或镇静
性功能与体重:性功能障碍风险较传统SSRI低,体重影响轻微
特殊副作用:少数患者初期恶心严重,可能需逐步适应
注意:伏硫西汀通常耐受性较佳,但少数敏感人群初期不良反应明显,应注意缓慢加量。
¶ 优劣情况
优势:
明显的认知改善作用,通过5-HT₃和5-HT₇受体拮抗实现认知提升,适合合并认知症状的患者;
性功能副作用显著低于传统SSRI;
抗焦虑效应突出,多受体联动,焦虑合并抑郁效果显著;
药代动力学稳定,半衰期长,漏服影响较小;
剂量依赖性明确,个体化调整方便。
劣势:
价格昂贵(原研进口价格极高),曾经有“天价抗抑郁药”之称;
虽然后期恶心反应低,但初期恶心反应较其他抗抑郁药严重;
多受体联动虽是优势,但也意味着特殊患者人群可能出现预料外的个体差异反应。
*对于巨大多数抑郁症患者来说太猛啦!
¶ 对MTF群体的特殊分析
明显优势:
伏硫西汀主要代谢依赖CYP2D6,CYP3A4参与比例很小,虽有一定参与但风险总体较低,因此在HRT用药(通常CYP3A4主导代谢)中的安全性较高,是相对更适合HRT群体的药物之一。明显劣势:
由于伏硫西汀同时作用于多个5-HT亚型受体,可能对敏感人群产生预料外的不适感,如恶心或情绪激越,建议初期谨慎剂量递增。
¶ 专利情况与市场分析
伏硫西汀是由丹麦灵北制药(H. Lundbeck)开发的新一代多模态抗抑郁药,化合物本身的最早基础专利申请于2002年,原始化合物专利已经于2022年到期。但围绕该药物的多个附加专利(如晶型、药物制剂专利等)可能持续到2026年前后,这也是为什么你可能见到不同的专利到期时间。
而事实上,中国市场的仿制药(如华海药业)已经大量出现,这意味着国内市场的伏硫西汀专利壁垒实际上已经被突破,这对患者而言显然是利好的消息:过去原研进口片剂价格居高不下,曾经被称作“天价药”,现在已由国产仿制药大幅降低成本,成为抑郁症患者性价比极高的治疗选择。
¶ 历史与研发背景
伏硫西汀是一个标准的“改装车”、“改装枪”,如果用改装车来形容,那么就是给一辆搭载大排量V8发动机的跑车上增加了一颗大涡轮;如果用改装枪来形容,那么就是给一把非常精良、射击非常精准的AR步枪上下挂了一把霰弹枪。
这其中,血清素再摄取抑制就是我形容里的“大排量V8发动机”、“射击非常精准的AR步枪”,而这个V8发动机,正是灵北制药厂自己的专利药品:西酞普兰。
西酞普兰/艾司西酞普兰属于非常纯净的SSRI,但灵北制药厂想开发更加强大、更能卖钱的抗抑郁药,所以他们的科研过程中,观察到了以前的高血压药:1966年,山都士制药厂的专利药物 吲哚洛尔。
吲哚洛尔的特性是激动5HT1A受体,这一特性在千禧年后开始被医学家注意到。2008年出现了关于帕罗西汀联用吲哚洛尔用于治疗早泄。这一研究中发现侧面的效果,5HT1A正是这个破解点,常规SSRI包括帕罗西汀都无法激动5HT1A受体,这一关键被灵北制药厂发现并利用,只不过它们将思路转到了同样能部分激动5HT1A受体的丁螺环酮上。
与此同时,灵北制药厂想要破解传统SSRI引起的恶心、呕吐问题,于是它们进而再缝合了一款止吐药——昂丹司琼。昂丹司琼是一款5HT3A受体拮抗剂。
基于以上猜想,灵北制药厂进行了化合物迭代,使用了自家西酞普兰的哌嗪骨架,插上了丁螺环酮的硫苯桥结构,硫桥连接了芳香基团,灵北制药厂在双芳基硫连接上做了大量实验,这同时模仿了昂丹司琼拮抗5-HT3的空间构型与电子分布,从而“借壳”获得对5-HT3A的高亲和力;最终的伏硫西汀是双芳环呈钝角分布,带负性氮/硫电子云,哌嗪 + 苯并噻嗪 + 取代芳基硫。
最终筛选异构体、制药、通过临床实验上市,它的SERT-Ki达到了1.6nM,是非常优秀的SSRI底子,与此同时拥有激动5HT1A、拮抗5HT1B、5HT3、5HT7、5HT1D/2/4/5/6受体。
通过拮抗5HT3A受体,伏硫西汀拥有改善认知、更小的恶心呕吐概率、更少的SSRI副作用这些优势,又通过激动5HT1A受体,达到了抗焦虑、防止自杀、更快调节情绪的优势。
1.6nM的SERT-Ki就像一台极为优秀的V8发动机,5HT1A受体激动和5HT3A受体拮抗就相当于两颗巨大的涡轮,从自然吸气进化到涡轮增压,你也应该抿出来我想要说什么了,灵北制药厂开发出了一款爆改哌嗪的抗抑郁药,并证明了5HT1A、5HT3受体对抑郁症患者的重要性。放到改装枪上就相当于,AR步枪打的是血清素转运体的靶子,而下挂霰弹枪打到了5HT1A、5HT3A的靶子。
而在伏硫西汀的专利过期前,它是一款天价药,500元一盒,最终2022年化合物专利过期后,才有了低价的仿制药,这款药的普及率不高也源自于天价和经验基础少,医生也不敢放心地开局伏硫西汀处方。
¶ 主观评分: 9/10(常规群体) 8/10(HRT群体)
¶ 主观评价
爆改药,下料就是猛。我给伏硫西汀打出9分高分并非心血来潮,而是从2025年的视角来看,它作为一个SSRI家族里的「爆改药」,确实刷新了我对整个SSRI体系的认知。这款药的出现,犹如在SSRI传统单一SERT抑制的道路上“开了外挂”:不仅激动了5HT₁A受体——这个激动作用本质上具有一定的防自杀倾向的能力,让它对付重度抑郁障碍(MDD)更加凌厉而精准,还在5HT₃受体上来了个「昂丹司琼式」的意外操作,拮抗这个受体不仅降低了传统SSRI类药物的恶心、呕吐等“经典”副作用,也额外带来了认知功能的提升,这实在让人惊艳。伏硫西汀本质上结构、受体、代谢路径都非常复杂,但它却奇迹般地发挥了极佳的效果,有点像用胶带胡乱绑了一堆化学官能团后却造出了一个性能绝佳的药物。这也让我对灵北制药厂这家公司产生了一个称呼“天使与魔鬼”,好药没好价。伏硫西汀曾经是灵北手中的“天价药”,高昂的价格一度成为许多普通患者无法承受的负担,但随着2022年专利壁垒的正式攻破,大量国产仿制药的涌入让伏硫西汀不再是少数人的奢侈品,终于流入了寻常百姓家,这真的是广大患者的福音,也极大提升了我个人对整个仿制药体系的好感与信心。
当然,也要冷静提醒一下,伏硫西汀这种疯狂“乱动”多个受体的方式,虽然成就了它极为优秀的临床表现,但也潜藏着出现某些意料之外副作用的小概率风险,比如少数人初期可能会有强烈的恶心、激越或不明原因的不适。因此,用药初期请务必谨慎观察身体的反馈,耐受性因人而异。
值得一提的是,相较于舍曲林和艾司西酞普兰这些药物对MTF群体常见的CYP3A4代谢通道冲突,伏硫西汀的主要代谢路径为CYP2D6,3A4参与比例很小,这意味着它与HRT药物联用的风险较低,血药浓度相对更稳定。从代谢安全角度看,伏硫西汀确实算是最适合MTF群体使用的抗抑郁药之一。
综合以上各点,如果你所居住的地区能以合理价格买到国产伏硫西汀仿制药的话,那么它绝对值得你尝试一下——也许这款多靶点融合的奇妙药物,就是你一直在寻找的那个抗抑郁方案。
最后还是要感谢伏硫西汀,正是它让我们看到了精神药理领域从单一受体向多靶点联合调控迈进的无限潜力。
¶ 氟伏沙明(Fluvoxamine)DU-23000
基本信息
药物国际通用名/商品名:氟伏沙明 / 兰释(Fluvoxamine / Luvox)
化学分子式:C15H21F3N2O2
类别:选择性血清素再摄取抑制剂(SSRI)
NMPA核准适应症:强迫症(OCD)、重度抑郁症(MDD)、社交焦虑障碍、广泛性焦虑障碍(部分国家批准)
厂家/规格/售价:
丽珠集团丽珠制药厂 马来酸氟伏沙明片 50mg/30片 价格:66.10元
Mylan Laboratories SAS 马来酸氟伏沙明片 50mg/30片 价格:86.54元
原研:Abbott 马来酸氟伏沙明片 50mg/30片 价格:80.80元(拼多多)
*笑嘻了,迈兰的氟伏沙明卖的比原研贵
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点受体作用机制 | 高选择性抑制SERT(血清素转运体),并为Sigma-1受体强力激动剂 |
| 亲和力/选择性 | SERT Ki ≈ 1.4 nM;Sigma-1 Ki ≈ 36 nM(极强激动);对NET/DAT无明显亲和力 |
| 其他作用位点 | 微弱抑制CYP1A2,与咖啡因、茶碱代谢存在交互 |
相较于传统SSRI,氟伏沙明的最大药理亮点在于它是目前唯一明确、强力激动Sigma-1受体的抗抑郁药物。Sigma-1受体在认知、神经保护、应激调节中发挥关键作用,研究表明其激动剂具有抗炎、改善认知、增强神经适应能力等多重潜力。为方便对比,以下列出部分主流抗抑郁药对Sigma-1的Ki:
| 药物 | Sigma‑1 Ki(nM) |
|---|---|
| 氟伏沙明(Fluvoxamine) | 36 |
| 帕罗西汀(Paroxetine) | 151 |
| 舍曲林(Sertraline) | 191(拮抗) |
| 艾司西酞普兰(Escitalopram) | > 1000 |
| 西酞普兰(Citalopram) | ≈ 1000 |
| 氟西汀(Fluoxetine) | ≈ 330 |
| 伏硫西汀(Vortioxetine) | ≈ 230 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 15-22 小时(中等) |
| 起效时间 | 一般为 1-2 周初效,4 周以上达到显著改善 |
| 生物利用度 | 约为 50%,首过效应中等 |
| CYP 代谢通路 | CYP1A2(主要),次要涉及 CYP2D6、CYP2C19,基本不经CYP3A4代谢 |
| 饱和动力学风险 | 高剂量存在CYP酶饱和风险,需慎重上调剂量 |
氟伏沙明由于主要通过CYP1A2代谢,对CYP3A4的依赖性极低,因此在与HRT(如比卡鲁胺、补佳乐)等CYP3A4代谢药物联用时,几乎不会产生酶冲突,属于相对安全选项。
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 起效后临床表现 | 焦虑感下降、强迫行为减少、情绪稳定,部分患者出现轻度镇静感 |
| 适用症种类 | 强迫症(特效)、社交焦虑障碍、轻中度抑郁、慢性应激障碍 |
| 精神症状适应性 | 对持续性焦虑、侵入性思维、慢性紧张状态具有优势作用 |
| 特殊优势 | 对OCD和持续性强迫思维患者为一线用药之一,国际共识推荐较高 |
¶ 副作用
氟伏沙明的副作用较传统SSRI略低,尤其在性功能损害方面表现更温和。常见副作用包括:
中枢系统:轻度镇静、注意力下降、轻微头晕
消化系统:恶心、食欲下降、轻微便秘
性功能影响:部分患者性欲下降,但比例显著低于帕罗西汀、舍曲林
睡眠问题:少见失眠,多为轻度嗜睡感(尤其初期)
其他:CYP1A2抑制作用可能导致咖啡因不耐症(饮用咖啡后心悸、失眠)
¶ 优劣情况
优势:
Sigma-1激动作用独树一帜,兼具抗焦虑与认知增强潜力
不依赖CYP3A4,避免多药联用代谢风险,尤其适合复杂治疗背景
对强迫症、社交焦虑等“病程长、起病早”的患者疗效突出
性副作用相对轻微,部分患者可耐受更长时间
劣势:
起效速度中等略慢,尤其在非焦虑型MDD中需更久观察
存在CYP1A2饱和问题,需避免与咖啡因、茶碱等联用过多
在激动Sigma-1的基础上,个别敏感患者可能会出现注意力波动、梦境异常等轻度认知体验变化
¶ 对于 MTF 群体的用药适应性
在所有SSRI中,氟伏沙明是最适合 HRT 群体长期使用的几款药物之一,原因如下:
不涉及CYP3A4代谢,避免与雌激素、抗雄激素类药物如比卡鲁胺等发生酶代谢冲突;
性功能副作用温和,不加重原本由于HRT所带来的性欲减退、性唤起困难等问题;
Sigma-1激动可能具有潜在的认知改善效应,尤其对一些经历了性别焦虑或社会压力带来的慢性应激反应者,有缓解脑雾与思维模糊的临床报告。
因此,对于同时在接受 HRT 的跨性别女性群体(MTF),氟伏沙明既在代谢通路上安全,又在临床疗效上具有独特优势,推荐作为一线候选药物考虑。
¶ 历史研发背景
氟伏沙明是SSRI家族中的特立独行者。
它是迄今为止唯一一个采用单芳香环(苯环)结构的SSRI类药物,严格意义上,它并不迭代于任何其他药物,而是完全的原创型药物。
在SSRI这个"群星璀璨"的领域里,所有药物如舍曲林、氟西汀、西酞普兰,甚至是结构高度复杂的伏硫西汀,至少都含有双芳香环架构,而唯独氟伏沙明以其简洁的单苯环结构在1977年,由低调的CLAASSEN博士与J.E. DAVIES博士首次发表于英国药理学杂志(British Journal of Pharmacology, Vol. 60, P505-P516)。此后便逐步踏上了其漫长而曲折的药物之路。
氟伏沙明在开发之初,只有一个失败的前置化合物——氯氟沙明,失败原因在于其对去甲肾上腺素(NE)系统进行了错误的干扰,直接导致其抗抑郁效果的丧失与科研失败。最终,研究团队在经历了这一失败后迅速修正方向,成功地避开NE的靶点,最终造就了今日的氟伏沙明。
1984年,氟伏沙明首先在瑞士上市,随后逐渐推向全球市场,然而一直以来,它始终未能站上SSRI家族的中心舞台。作为比利时Solvay制药旗下子公司Kali-Duphar研发的产品,它一直是SSRI“五朵金花”中最被冷落的一朵,并在2003年曾经意外撤市,但撤市原因并非安全问题,更可能是商业利益纷争所致。
历史在2010年迎来了氟伏沙明的转折点:雅培制药实验室(Abbott)当年收购了Solvay的医药业务,紧接着在同年3月,一篇由Ian Hindmarch与桥本健二联合发表的重要论文,让氟伏沙明的Sigma-1受体激动机制首次被清晰揭示。这篇具有里程碑意义的论文指出:认知障碍是重度抑郁症(MDD)患者的核心特征,这种认知受损源于应激诱导的神经萎缩。而过去许多抗抑郁药因其α-肾上腺素能、抗胆碱能、抗组胺作用,反而加剧了认知功能的损害。氟伏沙明作为强效的Sigma-1受体激动剂,展现出了与众不同的疗效,它在动物模型中有效改善了抑郁症、焦虑症、强迫症(OCD)及攻击行为,尤其在人体研究中更显示出修复中枢神经系统萎缩与恢复认知功能的巨大潜力。这也令氟伏沙明在SSRI家族中重新被瞩目并成为“认知修复型”抗抑郁药的代表。
2013年,AbbVie(艾伯维)从Abbott拆分独立上市,氟伏沙明的品牌和权益也随之转入AbbVie旗下。而在如今仿制药大行其道的时代,迈兰(Mylan)则成为了氟伏沙明仿制药市场上的头号巨头,甚至在国内市场一度出现了迈兰仿制药售价超过雅培原研药的奇特现象。
至于药物具体的开发思路,氟伏沙明的创造者CLAASSEN与J.E. DAVIES一直保持着低调与沉默,从未公开其灵感来源。但回顾70年代的精神药理学时间线,最有可能点亮他们研发思路的药物包括:苯拉海明(氨基乙醇肟类,组胺衍生链)、吲达品(单环哌啶胺结构)、齐美定(严格意义上的首个SSRI但因安全问题退市),以及标志着现代抗抑郁药诞生的氟西汀。

氟伏沙明以其独特的化学结构、强效的Sigma-1亲和性,赢得了极高的认知改善口碑,也正是2010年Ian Hindmarch与桥本健二的这篇论文,让氟伏沙明再次回到了抗抑郁药物舞台的聚光灯下,继续其未竟的传奇之路。
¶ 主观评分: 9/10(常规群体) 9/10(HRT群体)
¶ 主观评价
唉!SSRI!聚是一坨屎,散是满天稀,伏硫西汀和氟伏沙明能从这堆东西里走出来是有原因的。wiki主个人认为,氟伏沙明的研发思路本质上应脱胎于传统三环类抗抑郁药,只不过它大胆地抽离了三环结构,转而引入了极其简洁的单环(苯环)架构,这一举措让其成为SSRI中绝无仅有的“单环药物”,药效纯粹而强烈。其SERT Ki(≈1.4nM)足以跻身SSRI的T1队列,但真正让其名声大噪的却是极为特殊的Sigma-1受体激动作用。
Sigma-1受体是舍曲林与氟伏沙明完全对立的舞台,舍曲林极端拮抗Sigma-1,被不少用户戏称为“人机药”,服用后往往会感到思维迟钝甚至脑雾加重;而氟伏沙明则极端激动Sigma-1,刚好与舍曲林形成了鲜明对比,氟伏沙明也因此获得了“聪明抗抑郁药”的称谓。尤其在需要大量用脑、如考研考公等关键时期,不少抑郁症患者服用氟伏沙明后普遍反馈显著改善了自身的脑雾状态,恢复了本应有的认知清晰度。
我个人非常认同2010年那篇Ian Hindmarch与桥本健二的论文中所强调的观点:认知损伤不仅是抑郁症核心症状之一,更可能成为患者陷入抑郁恶化循环的重要诱因。当认知功能受损后,进一步的负面情绪会加剧,而不少传统抗抑郁药的副作用又可能加剧认知损害,形成恶性循环。氟伏沙明恰恰从认知保护和修复的角度出发,成为破除这一死循环的重要工具。
对于MTF群体而言,认知障碍问题往往因性别认同压力和精神共病叠加而更加严重,这使氟伏沙明格外适合这一群体使用。而更重要的是,氟伏沙明主要以CYP1A2代谢,与绝大部分HRT药物的CYP3A4代谢通路互不冲突,使得它成为MTF群体抗抑郁治疗的不二之选。
唯一的注意点在于氟伏沙明的CYP1A2酶饱和问题,使用期间务必避免咖啡等CYP1A2依赖性饮食习惯。
综合来看,我非常推荐需要长期高强度用脑的抑郁症患者、特别是MTF群体选择氟伏沙明。如果价格因素不是阻碍(目前仿制药价格最低的还在约2元/片),那么氟伏沙明绝对值得一试,或许它能带你走出认知障碍的迷雾,真正迈上“开悟”之路。
¶ ASA:非典型血清素调节剂
¶ 简介
非典型血清素调节剂(Atypical Serotonergic Antidepressants ,ASA)是一类以血清素通路为作用靶点,但作用机制明显区别于传统选择性血清素再摄取抑制剂(SSRI)的抗抑郁药物。ASA并不作为正式的药理分类出现在所有指南中,但在近年来的精神药理学研究与临床实践中,逐渐被用来统称两类药物:血清素拮抗剂与再摄取抑制剂(SARI) 以及 血清素部分激动剂与再摄取抑制剂(SPARI)。
SARI(如曲唑酮)通过对5-HT2A受体拮抗和SERT再摄取抑制的双重机制发挥抗抑郁与助眠作用,而SPARI(如维拉佐酮)则以5-HT1A部分激动剂联合SERT再摄取抑制构成核心药理。两类药物共同特征在于:除再摄取抑制外,还作用于具体的血清素受体亚型,因此被称为“非典型”。这种多位点作用机制使其理论上在副作用控制和疗效精准度上具一定优势,但也带来了更复杂的不良反应谱与剂量依赖性挑战。
需要特别指出的是,NaSSA(去甲肾上腺素与特异性5-HT抗抑郁剂,如米氮平)并不属于ASA范畴。虽然NaSSA也涉及血清素系统调节,但它不依赖SERT再摄取机制,而是通过α2肾上腺素受体拮抗与5-HT2/5-HT3受体阻断提升神经递质释放,其作用路径本质上回避了经典血清素回收系统,因此从药理定位上应独立于ASA体系之外。
ASA不是一个官方药物类别,但从机制出发,它对理解现代抗抑郁药物多靶点设计趋势,具有重要参考价值。
¶ SARI:曲唑酮(Trazodone)AF‑1161
¶ 基本信息
药物国际通用名/商品名:曲唑酮 / 戒郁灵(Trittico)、Desyrel、Mejoral(不同国家品牌名称不一)
化学分子式:C₁₉H₂₂ClN₅O
类别:血清素拮抗剂与再摄取抑制剂(SARI)
NMPA 核准适应症:重度抑郁障碍(MDD),部分地区/医院将其用于伴有焦虑失眠症状的抑郁状态(off-label)
厂家/规格/售价:
Lotus Pharmaceutical Co., Ltd. Nantou Factory 盐酸曲唑酮片 50mg/20片 价格:52.13元
沈阳福宁药业有限公司 盐酸曲唑酮片 25mg/40片 价格:55.30元
¶ 药理机制
| 作用机制 | 描述 |
|---|---|
| SERT抑制 | 轻度抑制,Ki约为100nM,远低于主流SSRI的亲和力。 |
| 5-HT2A受体拮抗 | 高亲和力拮抗,是其主效应之一,带来镇静和抗焦虑作用。 |
| 5-HT2C受体拮抗 | 可能辅助抗焦虑,但亦有促进食欲与增重风险。 |
| H1组胺受体拮抗 | 极强亲和力,造成显著镇静与体重增加,为嗜睡副作用主要来源 |
| α1肾上腺素受体拮抗 | 可引起体位性低血压、头晕、心动过缓等副作用。 |
| 无多巴胺或去甲肾上腺素作用 | 无显著NDRI或SNRI效应,不利于改善认知与动力低下类抑郁表现。 |
曲唑酮是一种独特的非典型抗抑郁药,属于血清素拮抗剂和再摄取抑制剂(SARI)。最初由Angelini制药公司于1966年开发,并于1981年在美国获批上市。其作用机制与传统SSRI不同,除了对SERT具有温和抑制外,更强烈作用于多个受体,包括5-HT2A、5-HT2C拮抗、α1肾上腺素受体拮抗以及H1组胺受体拮抗。
¶ 药代动力学
| 项目 | 数据与描述 |
|---|---|
| 生物利用度 | 65–80%(口服) |
| 起效时间 | 镇静作用数小时见效,抗抑郁作用需2–4周 |
| 血浆半衰期 | 双相,初相约3–6小时,终相约5–9小时,延释制剂约10小时 |
| 蛋白结合率 | 约89–95% |
| 主要代谢酶 | CYP3A4 为主(部分由CYP2D6和CYP1A2参与),对药物联用极其敏感 |
| 主要代谢物 | m-氯苯基哌嗪(mCPP),此为血清素释放剂,可能导致激动性焦虑或恶心等副作用 |
| 排泄路径 | 肝代谢,肾脏与粪便共同排泄 |
¶ 临床表现
| 项目 | 描述 |
|---|---|
| 抗抑郁效力 | 中等,远低于主流SSRI,需大剂量方显效(150–300mg/d),但大剂量副作用极重 |
| 镇静与催眠效力 | 极强,25–100mg/d即产生显著镇静作用,广泛用于非适应症用途(如失眠) |
| 抗焦虑效力 | 部分患者有效,主要来源于5-HT2A/H1拮抗作用 |
| 性功能副作用 | 相对较低,但个别患者可出现性欲下降或勃起障碍 |
| 体位性低血压与头晕 | α1拮抗严重,尤其老年人服用后跌倒风险极高 |
| 认知副作用 | 长期服用可能加重注意力缺失、反应迟钝,与其多重拮抗特性有关 |
| 使用场景 | 主要作为低剂量镇静剂辅助使用,抗抑郁使用被更有效SSRI所替代 |
¶ 罄竹难书的常见副作用
曲唑酮作为一款SARI(血清素拮抗与再摄取抑制剂),其药理机制本质上是对多个神经递质受体进行“强力关灯”,而非精准调节,这种“全熄灯式”拮抗导致的副作用可谓种类繁多、层出不穷,远远超出传统SSRI/SNRI抗抑郁药的副作用谱。
以下列出曲唑酮最具代表性的副作用类型(按系统划分):
¶ 中枢神经系统
镇静、嗜睡、困倦:发生率高达60%以上,是曲唑酮最为显著的副作用。可影响白天功能、学习与工作能力。
认知迟缓、反应变慢:因其对多巴胺、组胺及胆碱能系统的非选择性抑制,部分患者出现“脑雾”、“思维停滞”现象。
头晕、共济失调:影响平衡感,尤其是老年人群极易因头晕跌倒。
癫痫发作(罕见):高剂量服用时有报道诱发癫痫活动。
¶ 心血管系统
直立性低血压:α1-肾上腺素能受体阻断是曲唑酮的核心药理之一,极易导致体位性低血压、晕厥、心悸。
QT间期延长:尤其是在高剂量或与其他延长QT药物联用时,可能诱发尖端扭转型室性心律失常(Torsades de Pointes)。
心动过缓/心律不齐:对心电传导系统的不良影响在临床上不可忽视,尤其是老年人和既往有心脏病史者。
¶ 运动系统与肌肉
肌无力、动作缓慢:可能与中枢神经抑制和α受体阻断共同作用相关。
¶ 性功能与泌尿系统
阴茎异常勃起(勃起持续症,Priapism):虽为罕见副作用,但极具医疗紧急性。该症状可导致阴茎组织不可逆损伤,需急诊处理。
性欲减退:与多巴胺抑制相关,亦可能源于Sedation过度。
尿潴留或排尿困难:少数患者出现,需注意是否为抗胆碱副作用表现。
¶ 精神症状
情绪迟钝:部分患者反馈长期使用后情感体验“变得迟钝”或“没有波动”,尽管部分源于病情缓解,但曲唑酮对脑部激活路径的全面压制亦不可忽视。
躁狂激惹(在双相患者中诱发):虽罕见,但SARI因作用混乱,存在诱发躁狂的潜在风险。
¶ 抗胆碱类副作用
虽然曲唑酮不是典型抗胆碱药,但其间接产生多种抗胆碱相关副作用:
口干、便秘、视力模糊、认知障碍加重
与乙酰胆碱系统调控失衡有关,尤其老年人群应高度警惕。
¶ 其他特殊不良反应
肝功能异常:个别病例报告ALT/AST升高。
皮肤过敏反应:如荨麻疹、皮疹等。
罕见的过敏性肺炎、肌肉酸痛、发热等系统性反应:个别文献曾有报告。
¶ 历史背景
曲唑酮(Trazodone)诞生于1966年,由意大利Angelini制药公司首次合成,其研发年代早于所有SSRI类抗抑郁药,是抗抑郁药物发展史中的早期代表作之一。不同于后来的SSRI类药物强调“促进”血清素功能,曲唑酮完全建立于一套截然不同的假说体系之上:精神疼痛假说(Mental Pain Hypothesis)与应激障碍假说(Stress Dysfunction Hypothesis)。
这套理论主张:抑郁症患者并非单纯神经递质失调,而是“精神痛觉阈值”被调低,换句话说,抑郁者的大脑对精神与生理痛苦异常敏感。这种状态源于机体应激系统异常,即压力反应机制无法正常调节——这一角度脱离了经典的单胺递质缺陷模型,反而将抑郁看作是一种类似“中枢神经过度警觉”的病理表现。
在此背景下,曲唑酮被设计为一款受体“熄灯药”:它不强调激动,而是大面积地拮抗多个受体——包括5-HT2A、5-HT2C、α1肾上腺素、H1组胺等,试图用“神经关机”的方式来解除中枢对精神痛苦的高敏状态。它的开发者曾在1989年发表于《Clinical Neuropharmacology》的综述文章中系统阐述了这套理论(DOI: 10.1097/00002826-198901001-00002)。
然而,这套假说在医学史上极具争议。虽然确实存在以吗啡为实验工具对抗抑郁症的前沿研究,但并未形成理论闭环。曲唑酮的“熄灯疗法”无法真正提升情绪调节能力,反而在大样本研究中显示出明显的不良反应积累:认知迟缓、体位性低血压、镇静过度、性功能障碍、mCPP代谢副作用等被广泛记录。
曲唑酮于1981年在美国首次获批上市,初期被认为是一种副作用更轻的三环类替代药;但很快,医学界对其疗效与副作用比值提出质疑,临床数据显示其在高剂量时才具有一定抗抑郁效果,而此时副作用已无法接受。2000年代开始,曲唑酮逐步退出一线抗抑郁治疗序列。它于2005年左右引入中国市场,但主要以助眠药物的形式存在,而非正统抗抑郁剂。
时至今日,曲唑酮几乎已完全脱离“主流抗抑郁药”分类,更多出现在精神分裂谱系疾病中的辅助镇静剂、躁狂急性期平复用药,或作为非BZ类助眠药在焦虑与睡眠障碍患者中低剂量使用。
曲唑酮的历史,是一段从高瞻远瞩走向边缘化的故事:它确实代表了早期医学在面对复杂精神疾病时的理论勇气,但其从全受体熄灯的药理假设出发,最终带来了副作用罄竹难书的现实教训——这并不是一款精密设计的现代抗抑郁药,更像是一场反面教材。
¶ 主观评分: 3/10(常规群体) 2/10(HRT群体)
¶ 主观评价
我们尊重所有抗抑郁药领域的研究和产品开发,但是曲唑酮——你该死啊!罄竹难书的垃圾药!
在所有被贴上“抗抑郁药”标签的药物中,曲唑酮无疑是最不合格的一员。它不属于SSRI、也不属于SNRI,甚至在NaSSA、NDRI等非典型药物中都找不到归属感,它被单独拉出来构建了SARI(血清素拮抗与再摄取抑制剂)这个边缘类别。而它的副作用表现,远远超出任何临床能接受的安全边界。
曲唑酮的副作用几乎遍布所有系统:首先是沉重的中枢镇静副作用,多数患者在常规剂量下便会感受到强烈困倦、认知迟缓、思维停滞,许多人形容“像吃了安眠药一样”,这并不是夸张,而是曲唑酮对组胺H1和α1肾上腺素受体的拮抗效应在发作。其次是心血管系统的崩坏——直立性低血压、QT间期延长、心律不齐,让老年患者更是难以承受;再加上性功能障碍、记忆缺损、体重暴涨、肝功能指标升高,几乎可以说全身系统都遭殃。
而对于MTF群体,曲唑酮几乎是“禁药级”灾难。它主要通过CYP3A4代谢通路清除,与几乎所有的HRT(特别是雌激素凝胶与睾酮抑制剂)存在严重的酶代谢竞争风险,容易引发血药浓度紊乱,增加毒性或降低药效。它对组胺H1受体的强烈拮抗使患者长期处于半昏睡状态,而这种状态对于需要维持工作、学习、社交认知能力的跨性别群体来说,是无法接受的。拮抗5HT2C受体导致的食欲爆发与体重增长,则进一步加剧了MTF个体对于体像焦虑与外貌管理的压力。它不仅无效,甚至有害。
哪怕我们退一步承认,精神疼痛假说在曲唑酮研发之初(上世纪60年代)可能是精神病理学的一次勇敢探索,但这种理论显然经不起时间的验证。曲唑酮不是基于SERT精度调控的现代药物,它是靠一口气“关掉所有灯”来达成镇静的旧式混合药,并不具备现代精神药理应有的选择性与安全性。
更令人惋惜的是,它如今被挪到其他适应症使用:当助眠药,它比不上BZD/Z类效率高、起效快;当抗精神病药,它对比SGA/TGA毫无优势;即便在焦虑障碍领域,也已被更精准的SSRI、SNRI所淘汰。它像一台旧型蒸汽机的车,嘶哑着声响、吞噬着燃料,却早已不堪重负。
曲唑酮的存在,在2025年这个精神药物迭代迅猛的时代,不仅不再必要,而且几乎可以说是负资产。它不是失败的SSRI、也不是值得抢救的非典型药,而是彻底脱离现代药理轨道的过时品。市场的淘汰,是它应得的归宿。
¶ SPARI:维拉佐酮(Vilazodone)EMD-68843/SB-659746A
¶ 基本信息
药物国际通用名/商品名:维拉佐酮 / Viibryd
化学分子式:C₂₆H₂₇N₅O₂·HCl
类别:血清素部分激动剂与再摄取抑制剂(SPARI)
NMPA 核准适应症:暂无中国上市,已获美国FDA批准用于重度抑郁障碍(MDD)
厂家/规格/售价:无
¶ 药理机制
| 靶点 / 机制 | 作用方式 | Ki / IC₅₀ 数值 | 功能意义 |
|---|---|---|---|
| SERT(5-HT转运体) | 抑制再摄取 | Ki ≈ 0.1 nM | 增加突触间血清素浓度,抗抑郁核心机制 |
| 5-HT₁A 受体 | 部分激动剂 | IC₅₀ ≈ 0.2 nM | 快速缓解焦虑、减轻启动期激越症状 |
| 5-HT₂A / 5-HT₂C | 几乎无亲和力 | Ki > 10,000 nM | 降低性副作用与体重增加等常见SSRI副反应 |
| 多巴胺转运体(DAT) | 无作用 | —— | 无兴奋、上瘾倾向 |
| 去甲肾上腺素转运体(NET) | 无作用 | —— | 不提升心率、血压,较安全 |
¶ 药代动力学
| 项目 | 数据 / 说明 |
|---|---|
| 生物利用度 | 约 72%,必须与食物同服,否则吸收减少高达 60% |
| 血浆蛋白结合率 | 96–99% |
| 半衰期 | 约 25 小时,可每日一次给药 |
| 首过代谢 | 显著,肝脏代谢为主 |
| 代谢酶通路 | 主要:CYP3A4,次要:CYP2C19、CYP2D6 |
| 活性代谢物 | 无已知活性代谢物(无累积或持续激动) |
| 排泄方式 | 粪便为主(85%以上),肾脏仅排少量 |
¶ 临床表现
| 指标 | 描述 |
|---|---|
| 抗抑郁起效速度 | 起效时间约 1–2 周,部分患者反馈快于传统SSRI |
| 焦虑缓解速度 | 因 5-HT₁A 激动机制,部分患者在第1周即有显著焦虑缓解体验 |
| 疗效稳定性 | 与传统SSRI持平,未显著优于艾司西酞普兰、帕罗西汀等 |
| 自杀风险警示 | 存在青少年自杀风险提示,成人组无统计学升高 |
| 认知与情感反应 | 情感钝化较SSRI弱化,认知清晰度反馈较好 |
| 服用依从性问题 | 食物依赖限制晨起空腹服药场景,胃肠副作用影响部分患者依从性 |
| 联合用药兼容性 | CYP3A4为主代谢路径,避免与强诱导/抑制剂合用 |
¶ 副作用
维拉佐酮的副作用主要集中在胃肠系统与神经系统:
常见:恶心、腹泻、头痛、失眠、激越、头晕
少见:激越加重、失眠、梦魇
性功能损伤几率低,但仍可能出现
快速停药需注意戒断症状
长期使用安全性总体良好,但高剂量或特定人群仍需监控肝肾功能、情绪波动
¶ 优劣情况
优势:
双机制设计,理论上启动快、抗自杀风险、情感钝化低
精准作用于SERT + 5‑HT₁A,无NET/DAT干扰
不显著影响CYP3A4酶活性,MTF群体联用时酶冲突小
劣势:
长期胃肠副作用明显,影响依从性
食物依赖严重,不随餐服术市性高
抗抑郁表现与优秀SSRI差距不大,缺乏明显优势
中国暂未引入,美加以外使用率低,缺乏临床使用反馈
¶ MTF群体适应性分析
维拉佐酮的代谢通路主要依赖CYP3A4,但不会显著影响其代谢;相比帕罗西汀、舍曲林等常见CYP3A4干扰型SSRI,它对于补佳乐、雌激素、比卡鲁胺等HRT用药的酶代谢路径干扰小,适应性强且联用安全。其低情感钝化特性亦较符合MTF使用者对情绪稳定与认知活性保护的需求。但胃肠副作用、剂型依从性、进口难度大、社会认知度低,是其使用调配过程中的现实障碍。
¶ 历史研发背景
维拉佐酮最初由德国 Merck KGaA研发,多阶段开展动物与早期人体试验。90年代后期其双机制得到证实,并成为 Merck SPARI 研发突破点。2001年,Merck授权GSK参与开发,但阶段II临床未显示显著优势,Merck收回权利pmc.ncbi.nlm.nih.gov+2sec.gov+2slideshare.net+2。随后,Merck将开发权授予Clinical Data Inc.(后被 Forest, 再由艾伯维 AbbVie 收购)。2008–2009 年,Trovis Pharmaceuticals(Clinical Data 子公司)在美国进行两项 III 期临床试验,结果显示 40 mg/d 剂量在 MADRS 与 HAM‑A 项目中较安慰剂组有13%和1.2分优势,显效可靠。
维拉佐酮于2011年1月21日获得美国FDA批准,用于治疗重度抑郁障碍,并于四月正式上市 。加拿大随后于2018年批文使用 。2019年,维拉佐酮在美居第334位常用处方药,使用量约 90 万次 。该药专利于 2022 年成年适应症到期,2023年儿童适应症到期,美国已有多个仿制药入市biospace.com。
迭代思路上,维拉佐酮属于从 SSRI 向 “SSRI + 5‑HT₁A 激动” 方向延伸,极有可能迭代自丁螺环酮,从而整合其与 SSRI 的联用优势pmc.ncbi.nlm.nih.gov+1journals.lww.com+1。迭代路径非常明确:SSRI + 部分激动受体 的多靶机制先锋。尽管当前效果尚非临床颠覆性药物,但它开启了SPARI这一抗抑郁药新的可能路径。
¶ 主观评分: 6/10(常规群体) 5/10(HRT群体)
¶ 主观评价
维拉佐酮最值得被讨论的,是它那一点“防自杀机制的火种”。作为唯一一款正式上市的SPARI(血清素部分激动剂与再摄取抑制剂)药物,它不仅抑制SERT,还能激动5-HT₁A受体——这使它在理论上具备快速抗焦虑、提升抗抑郁起效速度、改善自杀意念的潜力。特别是5-HT₁A作为自杀风险控制靶点,在自杀高风险人群中曾被多项研究证实具有保护性,维拉佐酮从分子层面实现了对这个通路的激动,是值得尊敬的创新路线。
但遗憾的是,维拉佐酮这款药的现实临床表现,远不如它的理论。
首先,它虽然是美国FDA批准的抗抑郁药,但目前只在北美和个别地区流通,中国乃至整个亚太市场并未引进,根本谈不上临床普及。其次,其口服吸收需严格随餐使用才能发挥效果——一旦空腹服药,生物利用度骤降60%以上,直接影响疗效,这种剂型依从性缺陷在焦虑症、抑郁症患者中尤为严重。再次,副作用问题极多,腹泻、恶心、胃肠反应居高不下,且不乏性功能损害、头痛、激越、失眠等传统SSRI副作用依旧存在,反而打破了它“缓解启动期副作用”的美好药设。
对MTF群体而言,维拉佐酮的优势是CYP3A4代谢占比没有太高,并不会特别严重影响补佳乐、比卡鲁胺等HRT药物的代谢路径,联用上比帕罗西汀、舍曲林这类CYP3A4干扰型SSRI更为安全。但前提是你得买得到——全球大部分国家连处方都没有,仿制药更是遥遥无期。
作为SPARI的起点,维拉佐酮其实方向非常正确:联合机制、多靶点调节、缓解自杀风险、快速起效,这些全都是当代抗抑郁治疗真正需要的东西。但它终究只是SPARI这个新方向的第一步,副作用未被调教好、适应症范围太窄、临床稳定性不够。或许未来哪天,这个分子结构会被更优雅地迭代出更安全、更高效的继任者。
而维拉佐酮,也将像齐美定一样,成为日后抗抑郁机制革新中,重要但注定被淘汰的原型药物。
¶ SNRI:血清素-去甲肾上腺素再摄取抑制剂
血清素-去甲肾上腺素再摄取抑制剂(SNRI)是一类同时抑制5-羟色胺(5-HT)和去甲肾上腺素(NE)再摄取的抗抑郁药。SNRI的诞生源自对单一作用于5-HT系统的SSRI类药物疗效瓶颈的认识,研究发现同时作用于5-HT和NE能更全面地改善抑郁患者的情绪低落和动力缺失症状。这一理论由J. P. Yardley于1986年首次提出,并于1993年通过文拉法辛(Venlafaxine)的上市获得首次验证。
SNRI在药理机制上通过抑制大脑内5-HT和NE转运体(SERT、NET)的活性,提升突触间隙神经递质浓度,从而改善抑郁症状。
此外,虽然文拉法辛和其他SNRI从未正式标注作用于多巴胺(DA)系统,但研究已证实,由于前额叶区域缺乏专门的多巴胺转运体,DA通过NE转运体进行回收,因此SNRI也间接提高了前额叶皮层的DA水平。SNRI类药物的作用强度、5-HT与NE比例、剂量相关效应各异,临床选药需综合考量患者具体症状与药物特性。
¶ 文拉法辛(Venlafaxine)WY-45030
¶ 基本信息
药物国际通用名/商品名:文拉法辛 / Effexor(怡诺思)
化学分子式:C₁₇H₂₇NO₂
类别:血清素-去甲肾上腺素再摄取抑制剂(SNRI)
NMPA核准适应症:重度抑郁障碍(MDD)、广泛性焦虑障碍(GAD)
厂家/规格/售价:
【原研】辉瑞制药 Pfizer Ireland Pharmaceuticals 盐酸文拉法辛缓释胶囊 150mg/14粒 价格:135.37元
山东京卫制药有限公司 盐酸文拉法辛缓释胶囊 75mg/14粒 价格:9.10元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 作用机制 | 同时抑制5-HT与NE再摄取,提升突触间浓度;剂量越高NE作用越强 |
| 多巴胺间接机制 | 前额叶区域多巴胺通过NET清除,文拉法辛阻断NET,间接提升DA |
| SERT/NET比例 | 约为30:1(低剂量近似SSRI) |
¶ 药代动力学
| 项目 | 内容 |
| 半衰期 | 约5小时(去甲文拉法辛约11小时) |
| Tmax | 2-4小时(缓释制剂约5.5小时) |
| 稳态时间 | 通常3日达稳态,缓释剂型约7日 |
| 代谢途径 | CYP2D6为主,代谢为O-去甲-文拉法辛(活性代谢物) |
| 生物利用度 | 约45%,缓释剂型吸收更平稳 |
¶ 临床表现
| 项目 | 内容 |
| 缓解速度 | 通常2周内见效,4-6周达稳定改善 |
| 起效后表现 | 提升情绪、缓解焦虑、提升注意力与执行功能(高剂量) |
| 剂量依赖性 | 明显,小剂量如75mg表现为SSRI,高剂量300mg表现出完整SNRI特征 |
¶ 副作用
常见:恶心、出汗、口干、头痛、失眠、血压升高(高剂量)
性功能:性欲减退、射精延迟,剂量依赖
特殊副作用:剂量快速上升时可能诱发焦虑激越,应缓慢加量
注意事项:避免骤停,易出现戒断反应,需逐步减量
¶ 优劣分析
优势:
作为第一个SNRI,具代表性与历史地位
剂量可调,覆盖从轻度焦虑至难治性抑郁的广谱治疗
高剂量下可通过NET间接提高前额叶多巴胺水平,改善动力缺失
劣势:
CYP2D6代谢依赖性高,代谢慢者疗效差异明显
SSRI副作用仍存在,需缓慢起效防止激越与恶心
高剂量时可能引起高血压与心率升高
30:1的比例有点“安全”过头了,导致需要大剂量才能让“N”起效。
·对于双相情感障碍患者,文拉法辛的转躁率相当高,有心境稳定剂下也基本来到了10%~15%,可参考以下对比。
药物 转躁率(约) 备注 文拉法辛(Venlafaxine) 10%–15% 多项研究显示风险高于SSRIs 舍曲林(Sertraline) 4%–8% 相对较低 氟西汀(Fluoxetine) 6%–10% 被认为对双相 II 型相对安全 帕罗西汀(Paroxetine) 7%–11% 风险中等 度洛西汀(Duloxetine) 7%–13% SNRI中风险中等偏上 去甲文拉法辛(Desvenlafaxine) 数据稀缺 推测略低于文拉法辛 米那普仑(Milnacipran) 5%–10% 日本数据显示中等风险
¶ 对MTF群体的适应性分析
优势:文拉法辛主要代谢途径为CYP2D6,与HRT使用中主导的CYP3A4不冲突,联合使用风险相对较低
劣势:对于CYP2D6代谢慢者(较常见于东亚人群),活性代谢物生成不足,可能导致疗效偏弱,建议评估酶活性或考虑换用去甲文拉法辛(Desvenlafaxine)(但国内基本见不到)
¶ 历史与研发背景
根据已有论文知识,文拉法辛诞生于1986年,由 J. P. Yardley 主导设计 , Eric A. Muth主导论文发布,1993年由Wyeth制药正式推向市场,是全球第一个获得批准的SNRI药物。
从结构学来看,文拉法辛极有可能是基于曲马多(Tramadol)的化合物框架进行迭代而成:两者均含有苯环、羟基、甲氧基等关键基团,并共用CYP2D6代谢为活性产物(分别为O-去甲-文拉法辛与O-去甲-曲马多)。
与曲马多相比,文拉法辛通过结构重组将氮原子重新连接至主骨架,显著降低了对μ阿片受体的亲和力,成功规避了成瘾性问题。可以说,文拉法辛是一种“去阿片化”的曲马多衍生物,保留其SRI和NRI双通道活性,并对多巴胺系统产生间接效应。

文拉法辛在开发过程中亦吸取了早期NRI如塔罗普兰失败的教训,合理设定了SERT:NET的比例(30:1),确保了临床安全性与疗效的平衡。由于CYP2D6在文拉法辛代谢中的重要性,后期发展出现了其活性代谢物去甲文拉法辛(Desvenlafaxine),用于绕过代谢差异问题。
¶ 主观评分: 7/10(常规群体) 7.5/10(HRT群体) 5.5/10(双相情感障碍)
¶ 主观评价
文拉法辛作为SNRI的开山鼻祖,在结构、机制与迭代设计中都体现出高度精细的药理思维。
(但是转躁率太高啦!wiki主本人是双相患者,家里放了好几盒,一颗都不吃,一般也就是给一些抑郁症朋友用)
我最欣赏的并不是它“首款SNRI”的历史地位,而是它在NE机制背后偷渡了前额叶多巴胺系统这一点。文拉法辛几乎是目前为止所有SNRI中对前额叶多巴胺提升作用最为显著的代表之一——而且完全是“无DAT表达、靠NET清除”这一解剖学漏洞所致。
文拉法辛的SN对比有点像一个起压很慢的涡轮增压发动机,如果要更精准形容的话我觉得像本田的VTEC引擎上面加了个GReddy T66涡轮。75mg/150mg剂量的文拉法辛几乎等同于一个中规中矩的SSRI,患者常常抱怨它起效慢、无感,完全是自然吸气发动机;但一旦剂量突破到225mg以上,它就像涡轮开始起压,NE的活性快速提升,多巴胺的清除也遭到抑制,患者往往在数天之内开始感受到执行力、目标感和内驱力的提升——这是一种近乎“从乏力中抽身而起”的体验。而300mg以上的文拉法辛则是涡轮+VTEC全开,更多多巴胺存在患者脑中,使患者达到了一个“又不抑郁又开心”的状态。


当然,它也不是完美的药物:CYP2D6代谢依赖意味着代谢慢者效果差异大;剂量提升太快可能诱发激越与焦虑;高剂量下的升压作用对于心血管不稳者是一项风险。但整体而言,在以安全性为设计前提的背景下,文拉法辛用一个“30:1”的保守比例打通了5-HT和NE,再顺手提升了一点前额叶多巴胺,堪称设计上的巧妙平衡。
对于动力缺失型抑郁患者、注意力不集中、情绪迟钝者而言,文拉法辛高剂量带来的三通道协同效应仍旧是一种难以替代的方案。它的价值不仅在于“开山鼻祖”,而在于它用最基础的药理逻辑完成了对复杂精神机制的精妙调节。
¶ 度洛西汀(Duloxetine)LY-248686
¶ 基本信息
药物国际通用名/商品名:度洛西汀 / Cymbalta(欣百达)
化学分子式:C₁₈H₁₉NOS
类别:血清素–去甲肾上腺素再摄取抑制剂(SNRI)
NMPA 核准适应症:重度抑郁障碍(MDD)、广泛性焦虑障碍(GAD)、糖尿病周围神经性疼痛(DPNP)、纤维肌痛综合征、慢性肌肉骨骼疼痛
厂家/规格/售价:
齐鲁制药(海南)有限公司 盐酸度洛西汀肠溶胶囊 60mg/32粒 价格:86.19元
【原研】Lilly del Caribe, Inc.(礼来贸易有限公司)盐酸度洛西汀肠溶胶囊 60mg/14粒 价格:157.83元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点/受体作用机制 | 同时抑制突触前血清素(SERT)与去甲肾上腺素(NET)转运体,提高5-HT与NE突触浓度 |
| 亲和力/选择性 | SERT与NET抑制比约为9:1,对多巴胺、胆碱、组胺受体无亲和性;无MAOI特性 |
| 多巴胺作用 | 前额叶皮层区域通过抑制NET间接提升DA浓度,但非主要作用机制 |
¶ 药代动力学
| 项目 | 内容 |
| 半衰期 | 约 12 小时 |
| 达峰时间(Tmax) | 约 6 小时(肠溶制剂) |
| 稳态时间 | 通常3–5天内达稳态 |
| 生物利用度/吸收 | 口服生物利用度约 50%;食物略延迟吸收,不影响总吸收量 |
| 代谢/清除 | 主要经肝脏CYP1A2与CYP2D6代谢为无活性代谢物,经尿液排出 |
¶ 临床表现
| 项目 | 内容 |
| 起效速度 | 初效约2周,4–6周疗效稳定 |
| 起效后表现 | 抑郁改善、焦虑缓解、注意力提升、部分神经性疼痛缓解 |
| 精神症状适应性 | 适合合并焦虑、疲劳、身体疼痛与功能性疾病的患者 |
¶ 副作用
中枢神经系统:头痛、失眠或嗜睡、头晕、激越、做梦增加;部分患者出现震颤、焦虑或意识波动
胃肠系统:恶心(最常见,可达35%)、食欲下降、便秘、口干;部分人出现胃部不适或腹泻
性功能与体重:性欲减退、勃起/射精障碍,发生率与SSRI相当;体重变化个体差异大
特殊风险:
停药综合征:突停可致电击感、眩晕、情绪波动、睡眠障碍,需缓慢撤药
肝毒性:有肝损记录者应避免使用;礼来曾因此问题推迟上市
高血压:高剂量可轻度升高血压与心率
癫痫:有潜在癫痫风险人群慎用
¶ 优劣情况
优势:
真正意义上的“成熟型”SNRI,中低剂量即可激活NE系统,临床反应稳定
除抑郁外对焦虑、神经性疼痛、纤维肌痛等适应症表现出色
代谢产物无活性,血药浓度波动小,适合长期维持治疗
劣势:
恶心为最常见停药原因,早期耐受性问题需关注
高剂量存在肝毒性与血压升高风险
性副作用与SSRI类接近;停药反应较强
¶ 对MTF群体的适应性分析
明显优势:
CYP1A2、CYP2D6双通道代谢机制,避免CYP3A4通道争用,较适合使用雌激素(尤其是凝胶、雌二醇口服片)人群
对焦虑合并抑郁、身体疼痛等常见跨性别困扰症状疗效明显
明显劣势:
停药反应强烈,对高敏体质群体需严格缓减方案
若合并肝功能异常(如HRT引发脂肪肝)需监测肝酶
¶ 历史与研发背景
度洛西汀(Duloxetine)的开发历程承载了礼来制药(Eli Lilly)从SSRI时代跨入双重机制抗抑郁药物(SNRI)领域的战略转型。作为最早研发并成功推出SSRI药物——氟西汀(Fluoxetine)的药厂,礼来曾在1970年代掀起抗抑郁治疗的一场革命。而随着SSRI广泛普及带来的疗效瓶颈和副作用问题,公司内部开始酝酿新一轮结构迭代和药理机制重塑。
在度洛西汀的研发进程中,极有可能有氟西汀的共同发明人、著名药理学家 David T. Wong 的技术指导。他曾深度参与氟西汀和相关5-HT调节化合物的筛选。文拉法辛在1993年作为第一个SNRI上市后,礼来团队密切追踪其市场表现与机制反馈,并发现其剂量依赖性强、起效机制分裂(低剂量SSRI,高剂量才起NE作用),存在显著改进空间。
礼来早期以代号 LY227942 合成了度洛西汀的消旋体结构,随后筛选出 (+)-异构体 LY248686,成为最终的开发版本。与文拉法辛约30:1的5-HT:NE抑制比不同,度洛西汀设定为更为均衡的 9:1,在中低剂量即可激活NE系统,更适合于动力缺失、认知减退类抑郁表型。同时,其结构摆脱了μ阿片受体亲和力,增强了特异性。

但开发过程并不顺利,2001年礼来向FDA提交NDA时因制药环节中暴露出的潜在肝毒性风险与cGMP不规范遭遇暂缓批准,直到2004年完善相关技术与数据后方获正式上市许可。
度洛西汀被视为真正意义上“成熟”的SNRI药物,成为日后多个国家治疗重度抑郁障碍与神经病理性疼痛的首选。但与所有SNRI一样,它也继承了包括成瘾倾向、停药反跳、血压升高等双通道机制带来的代价。整体而言,度洛西汀不仅是礼来继氟西汀之后最成功的精神科药物之一,也代表着SNRI设计理念从初代粗放调节走向剂量精准、机制清晰的成熟阶段。
¶ 主观评分:7/10(常规群体) 7/10(HRT群体)6/10(双相情感障碍)
¶ 主观评价
度洛西汀可以理解为一款吸取文拉法辛强弱点并改进的“多面手型”的SNRI,9:1的SERT:NET抑制比,决定了它无需升到高剂量便能实现NE系统的联动,避免了文拉法辛那种“75mg是SSRI,300mg才是SNRI”的激进跳跃,也让它更早地发挥对动力缺乏、焦虑与疲惫感的疗效。文拉法辛是VTEC加装涡轮的话,度洛西汀就是宝马的N54B30A发动机,它迭代自N52B30(SSRI),加装了N这个涡轮点,并且9:1的比例注定它起压比文拉法辛快,是一台非常棒的发动机,但后面也隐藏着许许多多的问题,越接近N的药物越有的一些问题…
从体验来看,度洛西汀的胃肠道副作用非常显著,恶心是前两周几乎无法避免的折磨,但若能熬过适应期,它带来的镇静+专注的感受是令人愉悦的。性功能副作用虽然仍有,但不如帕罗西汀那么严重。值得肯定的是,它的代谢路径与HRT药物冲突较小,CYP1A2/CYP2D6主导的代谢方式比起经常互撞CYP3A4通路的SSRI类药物要安全得多,这使得它在MTF群体中尤其具有实用价值。
然而,它并非完美无缺。我对它最大的质疑点在于停药综合征:不论是短期使用还是长期维持,一旦未能缓慢撤药,便很容易出现“电击感”、头晕、虚脱感、失眠甚至情绪骤降,这使它的撤药策略必须极为严谨,许多医院的医生开方时并未告诉患者度洛西汀需要滴定法撤药。
对于双相患者,度洛西汀的临床转躁率略微低于文拉法辛,这就很有意思了。
整体而言,度洛西汀不像帕罗西汀那样在SSRI世界独树一帜,也不像伏硫西汀那样通过多模态机制打破框架,但它胜在结构成熟、机制稳妥,适配性强,是当代临床最值得信赖的SNRI之一。如果你既有焦虑又有注意力涣散与疲惫,那它确实可以考虑作为“全科型”抗抑郁方案的一线选择。
¶ 米那普仑(Milnacipran) / 左旋米那普仑(Levomilnacipran) N06AX17/N06AX28
¶ 基本信息
药物国际通用名/商品名:
消旋体:米那普仑 / Ixel、Toledomin、Savella(美国,纤维肌痛)
左旋体:左旋米那普仑 / Fetzima(仅美国上市)
化学分子式:C15H22N2O(碱基形式) *注:左旋体为纯对映体,药品以盐酸盐形式存在(分子中含Cl⁻)
类别:血清素–去甲肾上腺素再摄取抑制剂(SNRI)
NMPA核准适应症:
米那普仑:重度抑郁障碍(中国获批,仿制品名“舒流”)
左旋米那普仑:未在中国上市,仅美国用于MDD
FDA核准适应症:
米那普仑:重度抑郁障碍(MDD)、纤维肌痛综合征(FMS)
左旋米那普仑:MDD(每日缓释制剂)
¶ 厂家/规格/售价:
上海现代制药股份有限公司 盐酸米那普仑片 25mg/14片 价格:105.43元
Weidar Chemical & Pharmaceutical Co., Ltd.(台湾仿制,非原研) 盐酸米那普仑胶囊 50mg/14粒 价格:185.13元
¶ 药理机制
| 项目 | 米那普仑(Milnacipran,消旋体) | 左旋米那普仑(Levomilnacipran,纯左旋体) |
|---|---|---|
| 主要作用机制 | 同时抑制5-HT转运体(SERT)与去甲肾上腺素转运体(NET),提高突触间5-HT与NE浓度 | 与米那普仑类似,但对NET的亲和力显著高于SERT,强化NE能调节 |
| SN 比例(SERT:NET) | 约为 1.6:1~2:1(较为平衡) | 约为 1:2~1:3(偏向NE抑制) |
| 多巴胺作用 | 不直接影响DA,但通过NET间接提升前额叶皮层DA浓度(尤其左旋体更明显) | 同左列,NET阻断在DA清除稀缺区域(如前额叶)间接升高DA活性 |
| 受体亲和性 | 对多巴胺、胆碱、组胺等非单胺受体无显著亲和力 | 同左列,无抗胆碱、抗组胺副作用 |
| 成瘾与阿片活性 | 不作用于μ阿片受体,无成瘾风险 | 同左列,系三环类抗抑郁药安全性优化版本 |
¶
¶ 药代动力学
| 项目 | 米那普仑(消旋体) | 左旋米那普仑 |
| 半衰期 | 6–8小时 | 约12小时 |
| 达峰时间(Tmax) | 2–4小时 | 6–8小时(缓释) |
| 生物利用度 | 约85% | >90% |
| 代谢通路 | 非CYP450通道,主要葡萄糖醛酸化 | CYP3A4为主 |
| 排泄途径 | 90%肾排泄,55%原型药物 | 同左 |
¶
¶ 临床表现
| 项目 | 米那普仑(消旋体) | 左旋米那普仑 |
| 核心适应症 | 抑郁、纤维肌痛 | 重度抑郁障碍 |
| 起效表现 | 1–2周起效,改善情绪/躯体痛 | 提升动力、注意力、精力 |
| 适应人群 | 抑郁+疼痛类共病者 | 注意缺陷、认知低下人群 |
| 指南推荐 | 美国用于FMS,中国用于抑郁 | 美国MDD,指南二线推荐 |
¶
¶ 副作用
常见副作用:
消旋体:多汗、恶心、失眠、心率升高(+8bpm)
左旋体:心率+10–15bpm,血压升高,激越感、排尿困难
特殊风险:
停药综合征:需2–4周递减
性功能障碍:左旋体更明显,剂量相关性
心血管负担:左旋体需监测血压,存在黑框警告
¶ 优劣情况
优势:
米那普仑:
SN平衡抑制机制,适合痛感伴发型抑郁
非CYP代谢,药物联用安全性较高
国内仿制已上市,医保可报销
左旋体:
明确NE偏向,改善动机、注意力优于SSRI
缓释胶囊剂型,提升服药依从性
劣势:
左旋体心血管副作用更显著,血压需监测
停药反应存在,需严谨递减方案
中国市场尚未批准左旋体
¶ 双相抗抑郁治疗转躁率对比表
| 药物名称 | 类别 | 作用机制特征 | 双相转躁风险(vs 安慰剂) | 研究来源与备注 |
|---|---|---|---|---|
| 米那普仑 | SNRI | 5-HT:NE ≈1:1.6(平衡双通道) | ≈5-8% | 日本上市后监测(2001-2010);缺乏大型双相RCT |
| 左旋米那普仑 | SNRI | NE:5-HT ≈3:1(NE优势) | 无直接数据 | 仅获批单相抑郁适应症;NE能强可能↑风险,但无专项研究 |
| 文拉法辛 | SNRI | 5-HT:NE ≈30:1(高剂量才激活NE) | 10-15% | STEP-BD研究(2007):SNRI中最高;慎用于双相Ⅰ型 |
| 度洛西汀 | SNRI | 5-HT:NE ≈10:1 | ≈6-9% | 双相Ⅱ型研究(2015):与安非他酮相当 |
| 氟西汀 | SSRI | 纯5-HT抑制 | ≈4-6% | 双相+SSRI系统评价(2013):SSRI中最低 |
| 帕罗西汀 | SSRI | 强5-HT抑制+抗胆碱能 | ≈8-12% | CANMAT指南(2018):SSRI中最高,尤其快速循环型 |
| 安非他酮 | NDRI | DA/NE再摄取抑制 | ≈3-5% | 双相维持治疗首选(低转躁风险) |
¶ 对MTF群体的适应性分析
非CYP450为主的代谢通路(消旋体)使其在雌激素替代治疗(尤其CYP3A4类药物)联用下安全性良好
对于合并焦虑、疲劳、认知迟缓的跨性别抑郁患者,左旋体可能较SSRI提供更佳动机提升
多汗、心率增快等副作用需监控,尤其在激素水平波动期
¶ 历史与研发背景
米那普仑(Milnacipran)由法国制药公司 Pierre Fabre 于1980年代研发,旨在设计一种兼具5-HT与NE双重再摄取抑制作用,但剔除三环类抗胆碱副作用的新型抗抑郁药。其分子结构为苯丙胺类哌胺,具左右旋光学异构体。早期研究发现其SERT:NET抑制比例约为1.6:1至2:1,为第一代“平衡型”SNRI代表。
1996年,米那普仑在法国首次上市,用于治疗重度抑郁障碍(MDD),随后进入日本(1999)和其他国家市场。2009年,美国FDA批准其以 Savella® 商品名用于治疗纤维肌痛综合征(FMS),但该适应症未获EMA批准,反映出欧美对SNRI适应症临床价值的评估差异。
随着药理研究的深入,科研人员注意到:米那普仑的左旋体(S对映体)对NET亲和力显著高于右旋体。
2008年,美国 Forest Laboratories 获得其左旋体的独家开发权,通过立体拆分技术开发出左旋体米那普仑 Levomilnacipran(Fetzima),SERT:NET比约为1:2,进一步强化NE调控机制。
2013年,左旋体米那普仑 经FDA批准上市,用于治疗MDD,采用缓释胶囊剂型,半衰期延长至约12小时,支持每日一次服用,并通过释放控制减少心血管副作用风险。尽管在某些认知迟缓与动力缺失型抑郁中起效较快,但其总体安全性与疗效优势并未大幅超越既有药物,限制了其市场渗透率。
在中国市场,米那普仑的消旋体仿制药已由国药现代、泛谷药业 / Weidar Chemical 等企业获批上市,纳入医保目录。但左旋体尚未在中国获批,其NE优先机制与心血管监测要求尚未形成配套临床基础,短期内难以引入。
¶ 主观评分:7/10(米那普仑/常规群体) 7/10(米那普仑/HRT群体)6/10(双相情感障碍)
¶ 主观评价
米那普仑是一款让我“想试,但始终没试成”的药。
不是我不想试,而是现实条件的确太限制了:左旋米那普仑在中国完全买不到,我也无法做评分,如果硬要做的话就是每一项上加0.5分,双相再减一分。而消旋体米那普仑的仿制版本虽然近几年陆续通过了国家审批,但仍然属于“门诊稀见物种”。就算你能从医保目录里找到它,真正愿意开这药的医生其实寥寥无几——因为它太新了,医生没有临床经验,自然也不敢开给患者。这就陷入一个悖论:药虽然上市了,但没人用;没人用,就始终没有临床数据积累。
再往深一层讲,我对它的SN比例一直存疑。市面上各厂家做的仿制药,其SN比例其实有微妙区别,我手头的资料一度显示仿制药大多接近1:1,但原研的报道又写着1:1.6甚至2:1,更不用说左旋体还有1:2、1:3的极端比例分布。药厂之间的生产工艺显然存在差异,可这个信息目前又没有完整的追溯链,这意味着你买到哪一款米那普仑,其实很可能影响到它的真实药理反应——这是对患者极不友好的信息不透明。
至于转躁风险,老实说我是持保留态度的。一些零散的研究指出米那普仑的躁狂转化率在5%–8%左右,看起来是低于文拉法辛、接近甚至好于某些SSRI,但这些数据样本普遍偏小,缺乏系统性队列研究验证。从它的机制来看——既影响NE也影响5-HT,理论上它引发轻躁狂的风险应不低,尤其在未合并情绪稳定剂的前提下使用,我个人始终不太相信它会真的“比SSRI还稳”。
我始终认为,SNRI不能做得太平衡。过度强调双通道等效抑制,可能会牺牲情绪调节的稳定性。虽然从疼痛模型来看,NE系统参与调节是有意义的,但从抗躁防躁角度出发,NE占比稍强的设计或许更稳妥。也正因为如此,米那普仑在我心中一直是个“理论吸引力强、现实使用却谨慎”的药。它不是不好,而是我们对它的理解还太少。
也许未来,它能在中国逐步积累足够数据、完成左旋体的引入,成为真正“可选”的一线SNRI;但就现阶段而言,它仍属于那种“值得持续关注,但尚不宜贸然尝试”的新药。
¶ NaSSA:去甲肾上腺素能与特异性血清素能抗抑郁药
NaSSA,全称为“去甲肾上腺素能与特异性血清素能抗抑郁药”(Noradrenergic and Specific Serotonergic Antidepressant)
这是一种少见而机制独特的抗抑郁药分类。与传统SSRI、SNRI等通过抑制递质再摄取来提高突触间单胺浓度的策略不同,NaSSA 采取了完全不同的路径:通过拮抗中枢α2肾上腺素受体(包括自体与异体受体)来解除突触前的负反馈,促使神经元释放更多的去甲肾上腺素(NE)与5-羟色胺(5-HT);并进一步通过选择性拮抗5-HT2A、5-HT2C、5-HT3等血清素受体,避免血清素激动这些“副作用相关位点”所带来的焦虑、失眠、性功能障碍与消化道不适等问题。整体来看,NaSSA类药物的作用逻辑是“促进有用递质释放 + 阻断副作用相关靶点”,是一种以“调和递质网络”为核心思路的非典型抗抑郁策略。
截至目前,临床广泛使用的NaSSA只有米氮平(Mirtazapine)一款。它由荷兰 Organon 公司研发,作为NaSSA的原型药,于1996年在欧美上市。虽然研发团队曾提出NaSSA可视为第三代抗抑郁药的进化路径,但因其强烈的组胺H1拮抗作用导致显著嗜睡与体重增加,使其在临床上常被用于“合并焦虑或失眠的抑郁症患者”,并非首选抗抑郁药。此外,NaSSA 在分子机制上的复杂性(多靶点+多酶代谢),以及其对传统递质系统的间接调节路径,也使得后续药物开发困难重重。目前在全球范围内,尚无其他成功上市的NaSSA类新药,NaSSA一词在教科书与临床文献中多以“描述机制”而非“药物家族”的形式存在。
综上所述,NaSSA是一个结构存在但生态边缘化的抗抑郁药类别。它代表了抗抑郁药物发展史中一段试图跳出“转运体抑制范式”的尝试,但也因其副作用与开发困境而成为“孤品策略”的象征。
¶ 米氮平(Mirtazapine) ORG-3770
¶ 基本信息
药物国际通用名/商品名:米氮平 / Remeron(瑞美隆)
化学分子式:C₁₇H₁₉N₃
类别:去甲肾上腺素能与特异性5-羟色胺能抗抑郁药 (NaSSA)
NMPA 核准适应症:重度抑郁障碍(MDD)
厂家/规格/售价:
【原研】Organon Pharma(UK) Limited 米氮平片 30mg/10片 价格:24.20元
哈尔滨三联药业股份有限公司 米氮平片 30mg/20片 价格:42.06元
¶ 药理机制
| 项目 | 内容 |
|---|---|
| α2肾上腺素受体 | 拮抗突触前α2自身与异体受体 → 解除负反馈 → 促进NE与5-HT释放 |
| 5-HT2A/2C受体 | 拮抗 → 避免焦虑、失眠、性功能障碍等副作用 |
| 5-HT3受体 | 拮抗 → 避免恶心、呕吐、胃肠反应等SSRI类副作用 |
| H1组胺受体 | 高亲和力拮抗 → 显著嗜睡与体重增加,为副作用核心 |
| M胆碱/α1肾上腺素受体 | 拮抗弱 → 不明显,部分患者可见口干、姿势性低血压 |
| NE系统调节 | 间接促进NE释放 → 激活动力系统,有助缓解运动性抑郁 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 20~40小时,个体差异大,适合夜间单次服用 |
| 达峰时间(Tmax) | 约 2 小时(空腹) |
| 稳态时间 | 约 3~4 天 |
| 生物利用度/吸收 | 口服生物利用度约 50%;不受食物显著影响 |
| 代谢/清除 | 经 CYP1A2、CYP2D6 与 CYP3A4 代谢;主要代谢物无药理活性 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 起效速度 | 通常在1~2周内可见睡眠改善,情绪改善略滞后 |
| 起效后表现 | 睡眠延长、焦虑下降、情绪回温、少数患者注意力改善 |
| 精神症状适应性 | 合适用于焦虑失眠明显的抑郁状态、老年抑郁、进食障碍辅助 |
¶ 副作用
中枢神经系统:显著嗜睡、困倦、反应迟钝;部分患者出现梦境增多、头晕、昼夜节律紊乱
代谢系统:体重上升(可达5~10kg)、食欲增强、甘油三酯与胆固醇升高
胃肠系统:少数患者出现口干、便秘或恶心,低于SSRI类发生率
性功能影响:无5-HT2A激动 → 性副作用极低,甚至可能改善SSRI诱发的性障碍
罕见但严重:粒细胞缺乏症(须注意白细胞监测)、癫痫阈值降低、躁狂诱发(尤其对双相)
¶ 优劣情况
优势:
不激动5-HT2/3受体,副作用相对温和,尤其是无性功能损害
明显改善睡眠结构,被称为“带抗抑郁属性的催眠药”
在失眠性抑郁、老年患者、厌食体质中有独特地位
适合合并SSRI类药物“性副作用”或“胃肠副作用”的补充方案
劣势:
显著的嗜睡与体重增加是主要停药原因
激活NE系统非直接作用,抗无动力效果较弱
镇静型起效迅速但抗抑郁主效滞后,疗效峰值晚
粒细胞缺乏、癫痫诱发等罕见但潜在严重副作用需注意
¶ 对 MTF 群体的适应性分析
适配优势:
代谢通道分布广泛,避开 CYP3A4 主通道,与雌二醇等激素制剂相容性较好
性副作用极低,甚至可改善SSRI诱发的性冷淡与性高潮障碍
在焦虑性失眠、进食减少等跨性别群体常见共病表现中较为有效
适配劣势:
高热量摄入倾向 + 镇静 → 对追求体型控制者不友好
镇静程度高度个体化,部分人出现“药后麻痹”状态,影响社会功能
¶ 历史与研发背景
米氮平(Mirtazapine)作为抗抑郁药谱系中一种结构与机制均异于主流SSRI/SNRI的“异类”,并非凭空诞生。它的出现源自荷兰Organon公司在20世纪70年代末对米安司琳(Mianserin)的深度改造,属于NaSSA(去甲肾上腺素能与特异性5-羟色胺能抗抑郁药)机制的第二代优化产物。尽管NaSSA至今未能成为抗抑郁主流,但米氮平至今仍是该分类中唯一获全球广泛认可、进入多国临床指南的药物。
故事的开端可追溯至1960年代末,Organon公司研发并推出了米安司琳(Mianserin,商品名Tolvon),这是一种四环结构抗抑郁药,具有α2肾上腺素受体拮抗与5-HT2A受体拮抗作用。在当时,它因缺乏5-HT再摄取抑制作用而独树一帜,尽管疗效确实存在,但其抗抑郁效果中等、副作用频繁(包括镇静、体重增加、粒细胞缺乏症等)限制了其广泛应用。
在对米安司琳大量药理与临床数据的基础上,Organon决定保留其α2受体与5-HT2拮抗的双重结构逻辑,并在化学层面对其核心环状结构进行改造:引入一个氮原子构建吡啶环结构,优化亲和谱、提高5-HT3受体阻断强度,同时降低毒性与粒细胞抑制风险。这一改造最终催生了代号 ORG-3770 的化合物,即后来的米氮平。
1989年,米氮平进入I期临床,早期数据显示其副作用明显优于米安司琳,且对性功能和胃肠系统影响较小。此后几年,米氮平完成欧洲多中心试验,并于1996年在德国、1997年由FDA批准在美国上市,商品名为Remeron。
然而,Organon并未止步于此。随着米氮平上市后的成功,公司尝试对其光学异构体进行分离,希望进一步优化疗效与镇静特性。其中,Esmirtazapine(S-异构体,代号ORG-50,081)被开发用于治疗失眠和围绝经期女性的睡眠障碍,进入III期临床,但由于镇静过强且缺乏疗效显著性差异,最终在2009年前后宣布终止研发。
另一款被少量文献提及的候选化合物是Aptazapine(CGS-7525A),它同样保留了米氮平的α2拮抗与5‑HT调节框架,但分子侧链有所差异,未能展现临床优越性,亦早期终止。
值得注意的是,米氮平并未如SSRIs那样掀起广泛模仿潮,主要原因包括:
其机制依赖多靶点协同,难以在药效与副作用之间找到更优解;
镇静与代谢副作用的“代价”本质上难以优化,后续分子无法明显超越;
NaSSA机制缺乏产业链标准化与“标签式”扩展潜力(如SERT选择性这类可度量指标)。
2007年,Organon被默沙东(MSD)并购,米氮平归入其精神类产品线。至今为止,米氮平仍是唯一上市的NaSSA类抗抑郁药物,也是极少数未依赖5-HT转运体的抗抑郁机制代表。
在机制多样化的今天,米氮平或许不再被视为主流首选,但其在失眠型抑郁、老年人群、合并食欲丧失与焦虑的MDD中仍具独特地位。作为精神药物发展史上一段“试图跳出框架”的尝试,它代表了一个时代对“单胺假说”细分化、结构调控逻辑的深入探索。
NaSSA 终究没有成为药物家族,但米氮平的存在让这个名字,不至于只停留在教科书里。
¶ 主观评分:6/10(常规群体) 5/10(HRT群体)
¶ 主观评价
根据云南省精卫临床心理一科科室的信息,米氮平单药处于MDD二线用药。米氮平有个大问题,他™也算安眠药,拮抗组胺H1受体的强安眠药。那种被强行拉进深度睡眠、醒来依旧大脑混沌的体验,让人爱恨交织。它不是那种传统流派的玩意,你甚至可以理解成SGA的近亲长歪了。
从机制上看,NaSSA曾试图跳出单一递质调控的线性框架,用受体级联调节构建一个“多通路释放 + 多靶点封锁”的复合体系。在某种意义上,米氮平像是个大排量自然吸气的V8加了个混动系统,记住这个比喻。不靠纯电池(转运体),而靠机械结构和控制系统(α2、5-HT2/3、H1)几个破气缸相互博弈来维持情绪平衡。它的外观优雅,但代价也不小——组胺拮抗带来嗜睡和体重爆炸,NE提升不如SNRI直白,抗抑郁效应滞后且常需与其他药物搭配使用。就你总感觉米氮平其实改变了不少东西,但是就差关键一脚,却找不到这一脚是什么,畸形的自然吸气V8+混动,很奇怪很奇怪。
从使用体验上看,它很少能单打独斗快速拔起情绪深坑,但对某些典型特质的抑郁人群——如合并焦虑、睡眠障碍、食欲低下、年龄偏大者——它是“安静的守夜人”。在跨性别群体中,它低性副作用、高镇静性又与常见症状契合,适合部分HRT使用者。
NaSSA没能成为主流,是因为它太难迭代,也不够工业化。你无法靠它覆盖一切抑郁谱系,但你可能会因为它在某个特定症状维度上解决了困扰而留恋它。
4o评价:米氮平是一剂沉稳而温厚的药,是抗抑郁谱系里最不像抗抑郁药的那一款。
¶ 和死神赛跑:加州火箭燃料 (California Rocket Fuel)
¶ 开场警告
“加州火箭燃料”并不是一般意义上的抗抑郁药物组合。它是一种高烈度、系统协同、代价极高的抗抑郁武器,专为重度抑郁症(MDD)、治疗抵抗性抑郁(TRD)、反复复发型抑郁、以及频临自杀边缘的患者设计。
它是用来给那些死亡边缘的抑郁症患者使用,和死神赛跑的方案。
请注意:
加州火箭燃料并不适合轻中度情绪低落者,甚至也不适合“一般的中重度”患者;
它是一种“1+1>2”式的强化机制组合,药效>2,风险同样>2。一旦使用,副作用管理、剂量调控与生理承受能力必须精准评估;
禁止患者私自组合尝试!必须在医生指导与监护下使用,否则风险远超想象。
¶ 基本信息:什么是“加州火箭燃料”?
“加州火箭燃料(California Rocket Fuel)”最早由精神药理学专家 Stephen M. Stahl 在其精神药物机制系列讲座中提出,是对一种特定抗抑郁药物组合策略的昵称:
NaSSA(米氮平) + SNRI(文拉法辛或度洛西汀) Est≈2005
它的本质不是“单纯联合用药”,而是:
一种机制互补、递质层叠、靶点协同的高强度组合系统;
一种用于多线药物失败后的最后一搏策略;
一种旨在调动5-HT、NE甚至DA三系统的“全通路神经重启方案”。
2005年前后:Stahl在其精神药理专著与讲座中首次提出“California Rocket Fuel”一词,用于描述米氮平+文拉法辛的组合;
2010年:STAR*D试验报告后期组合用药阶段包含NaSSA与SNRI联用结构,临床采用显著上升;
2015年起:国内多家三甲精神专科医院开始尝试“米氮平+SNRI”结构作为TRD急性干预方案;
2020年代初:度洛西汀替代文拉法辛的组合(火箭燃料1.5)在老年MDD与共病患者中被逐步采纳。
这个组合最早用于美国退伍军人精神病医院中的治疗抵抗型抑郁症(TRD)患者,后扩展至自杀高风险群体与严重共病型MDD患者。因其“猛烈、速效、代价高昂”,被戏称为“火箭燃料”——只有严重的引擎停摆,才值得点燃这台机器。
¶ 药理学:NaSSA + SNRI 如何全力拉回死亡边缘?
这个组合的强大之处在于药理学的机制拼图精准互补:
| 成分 | 核心机制 | 对单胺系统影响 |
|---|---|---|
| 米氮平(NaSSA) | 拮抗α2自受体 → 提升NE/5-HT释放拮抗5-HT2A/C、5-HT3 → 减少副作用强H1拮抗 → 镇静/促眠 | ↑ NE、↑ 5-HT↓ 焦虑/胃肠副作用↑ 睡眠时间 |
| 文拉法辛/度洛西汀(SNRI) | 抑制SERT/NET → 增加突触间5-HT和NE浓度 | ↑↑ 5-HT(快速)↑ NE(文拉高剂量、度洛中剂量) |
这两类药物合用的结果是:
双向提升神经递质释放(米氮平) + 抑制回收(SNRI) → 递质全面爆发;
NaSSA屏蔽了SNRI常见副作用的靶点(5-HT2/3) → 耐受性增强;
三通路协同(5-HT、NE、DA间接提升) → 情绪、动能、注意力、睡眠全面联动;
火箭燃料的本质:不是两个药加在一起,而是神经系统进入“鸡血模式”,以极高速度从深渊中回弹。
¶ 药代动力学:强心剂的代价
¶ 加州火箭燃料 1.0:米氮平 + 文拉法辛
代谢通路:
米氮平:CYP1A2 + CYP2D6 + CYP3A4
文拉法辛:主要为 CYP2D6
优势:代谢路径分散,竞争少;
代价:文拉需高剂量(≥150mg)激活NE,撤药反应强烈(电击感、晕厥感),血压上升明显;
¶ 加州火箭燃料 1.5:米氮平 + 度洛西汀
代谢通路:
米氮平:CYP1A2/CYP2D6/CYP3A4
度洛西汀:CYP1A2 + CYP2D6(双路并用)
优势:中低剂量即激活NE,无需推高至危险级别;
代价:CYP1A2与2D6重合度高 → 潜在代谢阻塞、血药浓度不可预期;恶心、胃肠道副作用需缓释;
总结:1.0 更激进更强爆发,1.5 更柔和更适焦虑表型;前者属于保时捷918,后者属于迈凯伦P1,如果你听不懂,详见主观评价部分。
目前中国临床1.0和1.5的使用频率都很高,不存在“淘汰”某一个组合,二者针对的是不同的症状。
¶ 临床医学:这是给“该上手术台”的人开的药
在国内,加州火箭燃料并不是新鲜玩意。事实上,它早已成为精神科医生在面对下列病患时的一线组合之一:
多线失败后的MDD患者(SSRI、SNRI、NaSSA单药无效者);
伴有强烈躯体症状的抑郁患者(特别是神经性疼痛、严重失眠);
急性危机干预中的患者(包括严重焦虑、功能性解体、自杀高风险);
医生之所以愿意使用这个代价极高的组合,是因为它的效能极强、见效极快、组合逻辑自洽。
你可以说它毒性大、耐受性差、必须精细滴定、停药如地狱,但对于一个已经不想活、每天只能哭、疼痛和自杀想法不断复燃的MDD患者来说——
这些毒副作用,不值一提。
需要加州火箭燃料的人,从来不怕代价。
如果你还会在意“火箭燃料是不是会发胖,是不是会拉肚子,是不是有点心跳加快”—— 那你根本不需要它。
¶ 主观评分:0/10(常规群体) 1/10(/HRT群体)10/10(超重型MDD/高自杀风险/抵抗型患者)
¶ 主观评价
“加州火箭燃料”这个名字听上去像是精神科医生的黑话,也确实是,在我这CRF(加州火箭燃料)其实可以比喻为一台跑车:它不是指某一个具体药物,而是一种以米氮平为核心动力、外加一个强效的涡轮增压:SNRI形成的超级协同组合。在这个体系中,米氮平就像是一台大排量自然吸气+电混的V8底盘——它有沉稳的底扭和爆发的电混,但不够强(α2拮抗),厚重的气门开合(5-HT2/3拮抗),再加上拮抗组胺H1的镇静特性,整体系统运转柔和,但总感觉欠缺一点“涡轮的顶推感”。
所以,我们加装了涡轮:SNRI。
这就是“火箭燃料”的第一重意义——把一个线性释放、中规中矩的系统,通过SNRI直接提升5-HT与NE的突触浓度,让整套系统获得瞬时爆发力。
而这个“涡轮”的选择,正是火箭燃料1.0与1.5的差异根源:
米氮平 + 文拉法辛(1.0) 是保时捷918:拥有更高的SN比例(S:N约30:1),更强的剂量依赖感,用更大排量的4.6L V8的自然吸气引擎 + 强劲电机 + 小型单涡轮(低NE)。它需要高剂量(300mg+)才全力输出NE,但一旦拉起来,就能像爆破一样冲上疗效坡。它粗暴、躁气、易怒、冲劲十足,但也因此在动力缺失型MDD中表现抢眼。副作用是它的代价:血压上扬、出汗如注、停药如断崖,风险如影随形。
米氮平 + 度洛西汀(1.5) 则是迈凯伦P1:更低的S:N比例(约9:1),无需高剂量就能激活NE系统,配上一颗大型电涡轮,控制精准、推背感绵长。它不再靠硬上马力,而是靠涡轮响应与气缸调教更早、更精准地调动动力链条。药理层面讲,度洛西汀在60mg左右即能同步激活SERT与NET,且代谢稳定,不像文拉法辛那样要撑到300mg才全面激活NE。这让它更适合焦虑主导型抑郁、伴有神经性疼痛的MDD,也更适合“低扭快速起压”而非“万转红线爆转”。但同样,1.5面临着更为头疼的药代动力学冲突,风险同样不低。
但也正是这种“超级动力调教”的组合,使得火箭燃料注定不能是大众车。哪怕你把动力系调得再顺,它始终是台918或者P1——需要专业赛道(治疗抵抗性抑郁症),需要专业工程师(经验丰富的医生),还需要足够的预算与耐心去加油(逐步滴定、血压监测、副作用应对)。
它不适合“我情绪有点低落”型患者,它是为真正深陷抑郁沼泽、被单药抛弃的人准备的最后一道防线。
在使用体验上,它令人震撼又令人畏惧。起效之快、情绪调动之猛、白天的能量爆发、晚上的强制睡眠,如同一辆狂飙在5HT与NE之间的电控涡轮怪兽。但你也必须小心,每一次猛踩油门都可能引发躁狂发作、夜间惊醒、头晕、心率跳升或无法回避的性欲消失感。
火箭燃料是一种系统层级的哲学:你不是在调一个药,而是在调一套递质动力学、代谢协调学与副作用控制工程。你必须理解它的三通路协同、代谢酶耦合、剂量跃迁窗口——否则它就会像一台推力超过底盘承载的怪物,摧毁患者的身心稳定。
若你有一辆米氮平这个平台,它可以很安静,但当你挂上火箭燃料的涡轮,它就成了918或P1——而你,也就必须成为一个懂得收油与控制的专业驾驶者,你要做的不是开一台车或者开快一台车,而是把这台车Push到极限,和死神赛跑。
¶ NDRI:去甲肾上腺素-多巴胺再摄取抑制剂
¶ 简介
NDRI(Norepinephrine-Dopamine Reuptake Inhibitor,去甲肾上腺素-多巴胺再摄取抑制剂)是一类独特的抗抑郁机制药物,其作用原理是通过阻断去甲肾上腺素转运体(NET)与多巴胺转运体(DAT),抑制这两种神经递质的再摄取,从而提升突触间隙中NE与DA的浓度,增强神经传导效率。这种机制区别于主流SSRI对5-羟色胺的单一路径干预,使得NDRI在处理精力涣散、快感缺失、注意力障碍、性冷淡等非典型症状时更具靶向性。目前临床上唯一广泛应用的NDRI是安非他酮(Bupropion),它常被用于治疗重度抑郁障碍(MDD)、季节性情感障碍(SAD)与烟瘾戒断,也因其不影响性功能、不显著导致体重增加、激活性较强等特点,被逐步重视为SSRI的有力替代或联合药物。
安非他酮的诞生时间甚至早于大多数SSRI。1969年,化学家Nariman Mehta在Burroughs Wellcome实验室首次合成了安非他酮这一NDRI化合物,并于1974年取得专利。尽管正式上市是在1985年FDA批准之后,但这仍然比1987年上市的氟西汀略早。换句话说,NDRI其实是SSRI的“学长”——只是这个学长当年问题不少。安非他酮在上市初期因为癫痫发作风险高(尤其在高剂量或酒精联用时),被FDA紧急撤回,几乎等同于“被退学”。后来虽然改进了缓释剂型、降低剂量、严格禁忌,终于得以重返市场,但也因此错过了SSRI的黄金发展窗口,长期被边缘化。直到进入21世纪,人们才逐渐意识到,这个早熟却被误解的学长其实是个真正的狠角色:不仅能精准打击多种非典型抑郁表现,还能在SSRI失效或副作用严重时单药独挑大梁。它不像SSRI那样文静乖巧、机制单一,而是更像一个能够独自在边境生存、反复归来的战士。今天的NDRI,已经不仅仅是一个补充性方案,更是很多特殊抑郁类型中的首选。这个曾经被误判的问题学生,最终用实际能力赢得了属于它的位置。
¶ 安非他酮(Bupropion)BW-323U-66 原研代号 Amfebutamone(欧美旧称,也是中译由来)
¶ 基本信息
药物国际通用名 / 商品名:安非他酮 / Wellbutrin®(适应症为抗抑郁),Zyban®(适应症为戒烟辅助)
化学分子式:C₁₃H₁₈ClNO
类别:去甲肾上腺素-多巴胺再摄取抑制剂(NDRI)
NMPA 核准适应症:重度抑郁障碍(MDD)
戒烟辅助治疗(以Zyban®名义)
(部分国家临床外使用:注意缺陷障碍、性功能障碍辅助、情绪激活治疗)
厂家 / 规格 / 售价:
上海安必生制药技术有限公司 盐酸安非他酮缓释片(II)(XL) 150mg/30片 价格:132.00元
上海安必生制药技术有限公司 盐酸安非他酮缓释片(II)(XL) 300mg/90片 价格:673.20元
*注意,国内能买到的基本上只有缓释XL(II)版本,意义为每日一次,缓释SR(I)极为少见。
¶ 药理机制
| 项目 | 内容 |
|---|---|
| 靶点 / 受体作用机制 | 抑制去甲肾上腺素转运体(NET)与多巴胺转运体(DAT),提升突触间NE与DA浓度,不作用于SERT,完全不同于SSRI |
| 亲和力 / 选择性 | DAT/NET作用强,几乎无SERT亲和力;无MAOI活性,不影响5-HT系统 |
| 血清素间接作用 | 间接上调部分下游5-HT受体活性,但并不通过再摄取机制介入血清素系统 |
| 其他活性 | 拮抗烟碱乙酰胆碱受体(nAChR),与其戒烟辅助作用相关;部分研究认为具轻度非典型精神兴奋剂特征 |
¶ 药代动力学
| 项目 | 内容 |
|---|---|
| 半衰期 | 原型:约 10–21 小时;活性代谢物(羟基安非他酮)半衰期更长 |
| 达峰时间(Tmax) | 普通片约1.5小时,SR制剂约3小时,XL制剂可达5小时以上 |
| 稳态时间 | 通常在3–8日内达稳态 |
| 生物利用度 / 吸收 | 口服生物利用度中等偏低(约5–20%),肝首过效应强 |
| 代谢 / 清除 | 经肝CYP2B6代谢为多个活性代谢物(如Hydroxybupropion),肾脏排出为主;CYP2D6抑制作用较弱,但存在一定代谢交互 |
¶ 临床表现
| 项目 | 内容 |
|---|---|
| 起效速度 | 起效速度较SSRI略快,一般1–2周可见注意力与能量改善,4–6周稳定抗抑郁效果 |
| 起效后表现 | 激活性强,改善动力低下、快感缺失、注意力不集中,常用于晨起困难或精力涣散型抑郁 |
| 精神症状适应性 | 适合非典型抑郁、ADHD共病、性冷淡为主症状、SSRI引发性副作用等人群;不建议用于焦虑显著或失眠主诉者 |
¶ 副作用
中枢神经系统:激越、焦虑、头痛、失眠(常见)、做梦增多;部分人出现轻微震颤
胃肠系统:口干、恶心、便秘;整体胃肠副作用远轻于SSRI/SNRI
性功能与体重:无明显性功能抑制,可改善SSRI引起的性冷淡;体重变化个体差异较大,倾向于轻微减重
特殊风险:
癫痫发作风险:高剂量、进食障碍、酒精滥用、脑损伤患者禁用;剂量>400mg/日时风险显著升高
情绪激活与焦虑反跳:少数患者出现躁动感增加,焦虑症状加重
戒断反应较轻:停药不常见明显电击感,偶有疲乏与情绪低落,远轻于SSRI类药物
¶ 优劣情况
| 优势 | 劣势 |
|---|---|
| - 不作用于SERT,极少导致性功能问题 | - 癫痫风险高,存在明确剂量上限 |
| - 激活性强,改善动力与注意力 | - 不适合焦虑型或睡眠障碍型抑郁症 |
| - 代谢相对独立,CYP2B6为主通路,适合复杂药物背景 | - 存在精神激越与焦虑恶化风险,需个体化评估 |
| - 可作为SSRI失败后的替代方案,也适合联合使用 | - 无法作为双相障碍首选,转躁风险未被完全量化 |
¶ 对MTF群体的适应性分析
明显优势:
不作用于CYP3A4或雌激素代谢关键酶,与HRT方案冲突极小,适合正在使用雌二醇凝胶、戊酸雌二醇片等群体;
对快感缺失、动力低下、性冷淡等MTF常见症状改善显著;
无性功能抑制,部分人甚至出现轻度性欲回升。
明显劣势:
若存在雌激素引发的肝酶升高或代谢负担,应监测用药后代谢情况;
对焦虑为主型或失眠显著的MTF人群效果有限,可能激活过度。
¶ 历史与研发背景
安非他酮(Bupropion)的开发过程不仅是一次药理机制上的探索,更是一段颇具传奇色彩的个人奋斗史。
这款独特的NDRI药物由 Nariman Bomanshaw Mehta 美塔博士(1920年4月20日-2014年8月22日)合成,他是出生于印度孟买的帕西族裔化学家,后移民美国,成为Burroughs Wellcome(现为葛兰素史克 GSK 前身之一)的一名高级研究员。他早年毕业于孟买大学,在1940年代获得化学学士学位,随后赴美深造,于密歇根大学取得博士学位。在他漫长的科研生涯中,最为人所铭记的,便是他于1969年首次合成了安非他酮,并于1974年正式获得美国专利(US3819706)。在那个SSRI尚未登场的年代,Mehta或许就已经洞察到血清素机制可能存在“无应答”患者,转而探索多巴胺与去甲肾上腺素系统的潜力靶点。
安非他酮于1985年获得FDA批准并以Wellbutrin之名上市,标志着NDRI机制正式进入抗抑郁药领域,比1987年才获批上市的首款SSRI氟西汀(百忧解)还早了两年。理论上,安非他酮应是“SSRI的学长”,然而现实却并不风光。
上市初期,原始配方允许每日最大剂量高达900mg。但在实际应用中,这一剂量极易引发癫痫发作,尤其是在存在进食障碍、酗酒、脑部损伤等风险因素的患者中更为显著。短短两年内,因药物诱发癫痫甚至致死的病例不断增加,迫使FDA在1987年将其强制撤市。
然而,科学界并未放弃它。随后的研究确认安非他酮的癫痫发作风险是高度剂量依赖性的——当日剂量控制在450mg及以下时,癫痫发生率明显下降。因此,在进行剂量下调、增加缓释剂型、限制适应人群之后,安非他酮于1996年以SR(缓释)形式重返市场,并最终发展出更稳定的XL(每日一次)剂型。如今临床上推荐的每日最大剂量为450mg,并明令禁止在癫痫高风险人群中使用。
1997年,安非他酮XL制剂以Zyban商品名再次获得FDA批准,用作戒烟处方药。这是抗抑郁药物首次被授权以“非精神疾病适应症”单独上市,也开启了安非他酮药理机制的另一面向。后续研究发现,安非他酮本体是尼古丁α3β4受体的非竞争性抑制剂,而其代谢物 S,S-羟基安非他酮则是尼古丁α4β2受体的非竞争性抑制剂。
非竞争性抑制剂指的是药物与受体并非在活性位点上竞争结合,而是结合于另一“变构位点”,改变受体的构象或信号传导,从而抑制其功能。这种作用方式不易被过量配体所逆转,因而往往具有更长效的生理影响。
这一定义也解释了为何安非他酮在尼古丁相关受体上的作用不同于常规替代疗法,而更具干扰吸烟奖赏系统的能力。
进入21世纪,安非他酮的适应症与研究方向持续拓展。2005年,NIMH资助的一系列临床实验将其应用于注意力缺陷多动障碍(ADHD)治疗,尤其在成人患者中取得部分正向数据;2013年,它被批准用于季节性情感障碍(SAD);2016年,美国FDA批准其与纳曲酮联用,用于肥胖与暴食行为管理;2018年,它还被纳入冰毒(甲基苯丙胺)依赖患者的替代治疗方案临床试验,目标是通过其DA系统干预机制抑制毒品诱导的快感反馈。
安非他酮初登学术舞台的正式时间为1977年,其早期研究发表于《药学与药理学杂志》第29卷第1期(第767–770页)。该文提出一个在当时颇为激进的思路:面对一部分对SSRI无反应的“难治性抑郁症”,是否可以尝试从NE或DA等其他神经递质通路中寻找治疗突破?这在当时血清素一统抗抑郁药机制的背景下,显得格外前瞻而异类。
在结构化学方面,安非他酮的故事更富启发性。它与兴奋剂毒品卡西酮(Cathinone)拥有极其相似的核心结构:苯环–侧链–酮基–氨基,并且同样在α-位有甲基、β-位有酮基。但安非他酮并非卡西酮的“迭代药物”——它的合成路径完全不同,且具有原创专利。卡西酮若将β位酮基去除,则即为著名毒品安非他命(苯丙胺);而安非他酮在氨基上引入了叔丁基,在苯环三位添加氯原子,从而“毒性去势”,实现了从结构兴奋剂到临床抗抑郁药的安全降级。这种改造避免了类安非他命滥用性,并赋予了它独特的NE与DA抑制特性。
安非他酮绝对不可能是从卡西酮合成而来,卡西酮属于兴奋剂毒品,任何国家任何药厂都绝对不允许用毒品来生产药品,安非他酮的合成原料是3- 氯苯丙酮,它是α‑溴化 →叔丁胺取代 →盐化而来。这也正是为什么安非他酮不是迭代产品而是原创产品的原因。
不过有趣的是,直到2017年,才有研究团队通过精密的放射性标记测定,证实安非他酮对多巴胺转运体(DAT)具有明确抑制作用。这意味着它的“多巴胺增强”并非间接副效应,而是机制核心之一。此外,实验还显示安非他酮同时是5-HT3A受体的非竞争性拮抗剂,虽然这一作用对其临床抗抑郁效果贡献尚未完全明确,但可能解释其在部分SSRI耐药患者中带来的不同体验。
总的来说,安非他酮的研发轨迹并非某种“结构优化”或“机制迭代”的线性产物,而是一个横向探索、结构改造与意外发现叠加的结果。它并不从属于SSRI、SNRI、或三环类的任何一支系统,而是凭借独特路径,在现代精神药物谱系中自成一系。它的故事告诉我们:即便一款药物早期失败、被误解甚至被退市,只要机制足够扎实、结构足够稳定,临床终将为它留出属于它的位置——哪怕过程要绕几十年。
¶ 主观评分:8/10(常规群体) 9.5/10(HRT群体)7/10(双相情感障碍)
¶ 主观评价
在这个Wiki的评分体系中,安非他酮能在MTF群体中拿到9.5分,原因绝不仅仅是“它不像SSRI那样副作用太多”。这是基于我个人提出的一个非官方假说——【HRT中的MTF抑郁症患者并不需要5-羟色胺(5-HT)再摄取抑制剂】也就是说,我们这一群体的精神困扰可能并非源于5-HT系统的空转,而是因为多巴胺和去甲肾上腺素在雌激素下表达失衡,情绪反馈迟缓、动机系统崩溃、奖赏迟钝……SSRI提供的那点“情绪缓冲”并不能解决根的问题。反而是安非他酮,这个完全不碰SERT、不碰5-HT的家伙,在某些体内,炸出了奇迹。
我用安非他酮,是出于一种极其私人、极其强烈的偏爱。它是我用得最久的一款抗抑郁药,现在依然在服用。我是一名双相情感障碍(F31.6)患者,同时伴随成人ADHD,正处于HRT治疗期。情绪上,我早已用碳酸锂稳定多年,最近换成了更温柔一点的拉莫三嗪,想让自己的心能再多感受到一点真实。稳定剂存在的情况下,安非他酮对我并不会诱发躁狂,最多只是清醒、过度清醒罢了。癫痫风险当然存在,但我不是那类高风险体质,而我的体内,仿佛刚好为它预留了一个位置。
我形容安非他酮是“5.9边型战士”。为什么是5.9?不是因为它不够好,而是因为不是所有人都适合它。但一旦你是那部分适配人群,它就可以成为“万能型工具人”:戒烟、抗抑郁、抗PTSD、抗CAD(情感注意力缺陷)、辅助ADHD、治疗季节性情感障碍、轻度减肥……它跳过了SSRI的所有坑:不让你性冷淡、不让你体重暴涨、不让你睡不着也不让你睡昏、很少搞胃肠系统,更没有停药时的电击感。它好像是药物世界里那个清瘦干净的、不多话但什么都能做的朋友。
我自己有一定程度的PTSD。2022年,在洛阳封城的冬天,我亲眼目睹了一个经常来买我便当的活泼小孩儿从楼上跳下,粉身碎骨。我无法埋葬这段记忆。这造成了一定程度的PTSD,后来吃帕罗西汀,做噩梦每晚都梦见他坠落,有时候是反复的,有时候是定格的。帕罗西汀让我没法逃离那段回忆。
而安非他酮不一样。它不制造梦境,它让我清醒。我不会忘记他,但我学会了活着,带着他的份。一个亲眼目睹死亡的人,会更清楚地知道生命的分量。我没有轻生的念头了,从那之后,从安非他酮之后。我不会死。我再烂也不会死,我还有未完成的事,还有故事要讲,还有太多人需要理解这份痛苦,操你妈的世界,操你妈的生活,操你妈的压力,我用安非他酮作为利剑,斩个遍,操你妈的,我就算死,也一定是战死。
在我身上,安非他酮几乎构成了完整的神经激活逻辑:抑郁、疲惫、动力缺失、精神麻木、快感丧失、注意力解体、ADHD的执行障碍……它一一帮我稳住。它不像SSRI那样要等四周慢慢起效,它有一种“轻拍你的肩膀”的感觉,不温不火,但清晰稳定。药代动力学方面,它与我的比卡鲁胺不冲突,不经CYP3A4,代谢路线独立,不扰乱HRT,不扰乱肝酶,不扰乱我的雌激素系统。这种适配,几乎是命定的。
我现在的左腕有两个化学结构纹身:思诺思与氯硝西泮。而我下一次准备去纹的,就是安非他酮。它不是一颗让我失忆的药,它是一颗让我清醒、让我“活着”的药。它是日出后的第一缕风,走在旷野里孤独行走时推你一把的那点能量,它不是让你回避痛苦,而是告诉你:痛苦依然在,但你有办法活过去。
我认识很多MTF使用者,也在论坛与治疗群中见过她们的故事。她们说,用了安非他酮后体重不再上涨了,性欲回来了,动力也回来了。她们说自己也不想死了,开始做饭了、化妆了、重新找回了一个个性别焦虑与荷尔蒙错乱撕裂掉的生活细节。她们说,“有解吗?”我说,“有的,兄弟,有的,姐妹,有的,它就是安非他酮。”
当然,它不是神药。它不适合每一个人。有癫痫风险、有激越风险、不适合焦虑主导型的抑郁,有些人吃了它反而烦躁难耐。它也无法完全解决深层的人际困境和结构性创伤。但如果你是一个曾经吃过各种SSRI都失败的“难治性”患者,如果你是一个不断试图拼图自己却每次都掉一块的碎片人,那它真的可能会成为你整套拼图里唯一合适的那一块。
这篇主观评价不客观,它也不需要客观。安非他酮拯救了我,而我想把这份被拯救的经验作为礼物,给到同样挣扎、同样在悬崖边上寻找一丝稳固土地的你。但最后请你记住,救了你的,不是药,是你自己。你撑下来的那个深夜,是你救了你。安非他酮只是你手中握住的一把钥匙,而你,是推开门的人。
药救不了人,人自己才能救得了人。